In Did Pompe Disease Get a New Champion in President Trump?, I discussed the significance such a spotlight as a presidential address in front of Congress and the world can have on so rare a disease. Despite the fact these types of “Orphan Diseases” individually impact small populations, collectively they affect 25 million Americans.
Now, we will dive into why they are so important to understand, raise awareness about and divest resources for in order to advance therapeutic and potentially curative research.
Inborn Errors of Metabolism reflect a host of rare, genetic disorders that can be devastating, fatal and, in certain instances, if not swiftly detected in early infancy lead to catastrophic impairments in normal development. When we think of metabolism, we often think of our ability to rev ours up to lose weight or blame it for the reason we can’t. Here, we think of aesthetics and vanity. With these conditions, the end result is way more destructive and substantial than that. This complex constellation of diseases more specifically reflects when metabolism is profoundly influenced on a cellular level causing food not to be used effectively as an energy source.
A metabolic disease involves a defect prompting an abnormal chemical reaction throughout the body’s cells. This serves to prevent our ability to break down food that would otherwise be converted to energy to nurture our brains, enhance mobility, effectively pump our hearts and allow us to thrive and develop.
Think biochemistry and the urea cycle. Ring a bell? These diverse and complicated illnesses include organic acidemias, disturbances in fatty acid oxidation, lipid storage or amino acid metabolism, to name a few. Symptoms arise in a wide array. The biggest clues that point doctors to consider these conditions include when an infant is suddenly lethargic (which is not simply sleepy, but genuinely difficult to arouse), vomits, breathes rapidly, is persistently irritable, has seizures and moments of not breathing (aka apnea).
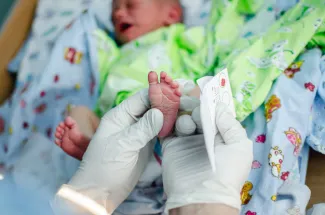
Fortunately, with the advent of the newborn screening program, there are a number of ailments in this category detected in the early weeks after birth. But, not all. This program entails taking a sampling of blood of all babies after 24 hours of life while still in the hospital. The specimen is collected from a heel prick onto a special filter paper that is sent to your respective state laboratory where it is tested for a whole host of conditions—many of which do quite well if intervention occurs quickly.
This testing began when it was realized many decades ago that a large number of people institutionalized for profound mental disability really had a disease called PKU (aka phenylketonuria). The sad reality was that had the PKU been identified very young then all of those patients would have developed normal intelligence and mentation. PKU patients lack an enzyme that breaks down the amino acid phenylalanine. As a result, when they ingest it, it deposits in the brain in excess leading to mental retardation and delays in development. An easy dietary shift to low phenylalanine intake— with a special formula as a baby— yields normal development. Thus, normal growth into adulthood. Ostensibly, a normal life.
This is why the newborn screen blood test is often called the PKU test. Over the years, many diseases that involve this kind of story have been added to it and the exact illnesses tested for varies somewhat between states. HIV, congenital hypothyroidism and sickle cell disease are a part of this testing—examples that are not metabolic disorders.
The newborn screen allows doctors to change the course of a patient’s life. Often the conditions screened don’t show signs until the damage has been done, so it allows for early intervention and truly is life changing for those affected. Not all metabolic disorders are covered in this sample, so expanded testing by the pediatrician is sometimes necessary—often arising from clinical symptoms. Adding other conditions to the official newborn screening test offered by your state involves a rigorous review process and approval.
Glycogen Storage Disease Type II is the umbrella term under which the inherited and often fatal Pompe Disease falls. Pompe disease results from a lysosomal deficiency in acid alpha-glucosidase (GAA) so the body is unable to break down the complex sugar called glycogen. Glycogen is the storage form of sugar (aka glucose). These patients have little or no activity of the GAA enzyme. That resulting buildup of glycogen, especially in muscles, prevents them from functioning normally —especially in the heart, skeletal muscle and other tissues.
Their hearts can enlarge and thicken without essential therapies. There are many types of Glycogen Storage Diseases influenced by which enzyme or sets of enzymes are problematic. Some forms can adversely trouble the liver more while others the brain, nervous system and heart. There are varying degrees of disease severity depending on which type. Certain types with proper treatment can result in normal lives, but not all. The ones like Type II are extremely challenging because they injure multiple organ systems and, therefore, can be fatal. Type II also presents differently depending on which form since the variants can appear at different ages. Breathing issues, cardiac abnormalities, unsteady gait and muscle weakness, difficulty swallowing (putting at risk for aspiration) and lung infections are among the innumerable hardships patients face.
Fortunately, most major academic tertiary care centers have medical genetics departments and each state has designated metabolic centers so detection and treatment can be implemented. To those families directly impacted and for future prevention, there is so much more work that needs to be done. Concerns over inheritable conditions should be readily discussed with your doctor.



