By Ruth Kava —
Scientists report they have successfully treated hemophilia B by giving sufferers gene therapy. When they infuse a gene for the correct blood-clotting factor, it's taken up by the patient who then can produce it on their own. So they no longer have to inject clotting factor to avoid potentially crippling or fatal consequences.
Hemophilia is a genetic disorder that impairs the ability of blood to clot. Obviously, this can be life-threatening in the case of an injury, but it can also cause excruciating pain and severe damage when internal bleeding damages joints simply because of routine wear and tear. Blood clotting is a complex phenomenon, involving a cascade of numerous proteins called clotting factors; the lack of any of these factors can impede the process.
There are two main types of hemophilia, A and B. Hemophilia A is the most common, seen in about 1 in 5000 live births. Type B type is much rarer, occurring in about 1 in 20,000 to 30,000 live births. Hemophilia A results from a gene mutation that causes the body to make less (or even none) or an abnormal type of clotting factor called (CF) VIII, while sufferers of hemophilia B (also called Christmas disease) have a mutation that similarly affects factor IX.
Currently, both types can be effectively treated with the infusion of the missing factor — made either from human plasma or from factors synthesized via genetic engineering. The main problems with this treatment are that it must be repeated for the person's lifetime (maybe as often as 3 times per week), and it is possible that the body will form antibodies (also called inhibitors) against the factor, which will decrease its effectiveness.
However, a more effective way to treat the disease would be by substituting a functional gene for the mutated one, which would allow the body to make normal amounts of normal CF and eliminate the need for the injected version. And researchers have recently reported that they have successfully managed to do just that in people suffering from hemophilia B.
Dr. Lindsy A. George of the Children's Hospital of Philadelphia and colleagues recruited 10 men with hemophilia B who produced 2 percent or less of the normal activity of CF IX. They all used CF IX either as needed to stop bleeding or as prophylaxis.
The scientists produced a bioengineered viral vector in a capsid from hepatic (liver cells) that contained a normal version of the factor IX gene. This was infused into the 10 patients with hemophilia B, and their levels of normal CF were followed for over a year. Before the infusion, the bleeding rate among all the participants was 11 bleeding events per year (range 0 to 48). But after the infusion, that rate dropped significantly to 0.4 events per year. This decrease is even more impressive given that the patients who had used factor IX prophylactically no longer did so, and still lowered their bleeding rates. Thus the activity of the infused correct gene was not only effective but was long-lasting, as shown in the graph below.
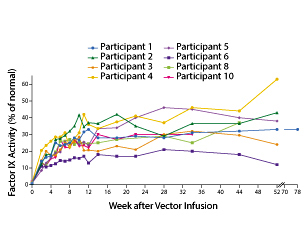
The authors noted "the mean steady-state factor IX coagulant activity was 33.7±18.5% of the normal value, and a therapeutic effect at this dose was observed in all the participants, even those who had preexisting (low titer) neutralizing antibodies or cellular immune responses that occurred after infusion. "
Of course, this was just a small study to demonstrate efficacy and safety of gene therapy for this particular condition. But if these results are replicated by other investigators and in larger studies, and if such bioengineered genes can be made available at a reasonable cost, we may well have seen the beginning of the end of hemophilia B, and hope for other similar diseases.
Category