Donald Trump. The Pharmaceutical Industry. If those terms two don't get you into a fight at a cocktail party, nothing will.
But, they need to be discussed, because there could be some big changes coming in both the manner and speed with which new drugs navigate the arduous US Food and Drug Administration pathway from lab bench to pharmacy.
Just one example to provide some context. Media have been making much of the fact that Jim O’Neill, a libertarian with a free-market mindset, has been floated as one possible candidate to head the FDA. Some of his prior statements are troubling but his philosophy could also be helpful, since he is "pro-biotech."
Either way, in talking about one even possible person we can flesh out what qualities we want in a new FDA leader, a body that is simultaneously criticized by activists on one side as rushing products to market, while the other side notes that approval costs and time have skyrocketed and become a barrier to small companies helping the public.
How can the FDA be both at the same time, and what would be needed to fix it? Let's take a look.
Drug Safety:
Here's the concern about just breaking the existing system. In the keynote speech in the Rejuvenation Biotechnology 2014 conference, Mr. O’Neill spoke about letting drugs be approved once they are considered safe, and that efficacy data would follow: "Let’s prove efficacy after they’ve been legalized.”
That is an obvious worry for people like me with 27 years of drug discovery experience. There can be no such thing as approval based on being either "safe" or "effective" because neither can be taken alone. Both factors must be considered or it is impossible to know whether the experimental drug can work at a dose that does not impart unacceptable side effects. It is the comparison of risk and benefit determines the feasibility of any given drug. In the absence of either of these, there is nothing to compare the other to.
For example, a typical dose of Prozac is 20 mg. It usually works fairly well, (1) and the side effects, while sometimes unpleasant, are rarely serious. But, at a dose of 2 mg it won't work at all, and at 2,000 mg death is likely. Given just this information, Prozac could never be approved as a drug. In other words, without dose ranging studies, which measure efficacy as a function of dose, the chances that the correct dose of the drug will be used would be slim, making the risk unacceptable.
Prozac demonstrates this even better in real world use. The recommended dose range is 20-60 mg per day. Higher doses not only do not work any better, but they produce significantly more side effects. So, while Prozac may be found to be safe at a two mg dose and deadly at a 2,000 mg dose, without determining the correct dose - not just the safe one - people would inevitably end up taking either too little or too much of the drug, which are both dangerous situations.
Drugs cannot be approved based on safety data alone, except in certain cases, such as untreatable terminal cancers or fatal disease for which there is no treatment at all.
Efficacy data:
Can the consumer market really determine efficacy? In other words, if dosing and safety are established, is it reasonable to just let people try it and see if it has benefit?
It cannot. Presumably, efficacy data would be obtained by reports from doctors who prescribed the drug, but this is unrealistic. Let's say that a doctor prescribes a drug under these circumstances. Here are the obstacles he or she will face:
- The doctor will not know for certain what dose to give, and may be reluctant to give the drug at all.
- The doctor will be writing a very small number of prescriptions compared to the number of patients that are studied in efficacy trials, so any effects gathered on subsequent visits will be essentially meaningless because of such a small dataset.This is especially true now, given the limited time of doctor-patient visits.
- The patient could die or become ill from the drug.
- The patient could die or become ill because the drug didn't work.
- The patient could die or become ill for reasons unrelated to the drug.
When taken together, it becomes clear that under such a set of 'efficacy after approval' rules, not only will we have less safe drugs, but it will take far longer, if ever, to determine whether they work or not. A measure such as this would slow new drug discovery, not expedite it.
But it's not all bad news for someone who wants to shake up an FDA process that now takes twice as long and costs twice as much as it did a generation ago.
One way of improving pharmaceutical innovation is to enable small companies to succeed. Right now, they often can't because of the government regulatory process. So someone with understanding of the discovery process could help promote the development of important novel drugs.
Biotech:
Since most startup drug companies are biotechs, which face a constant battle to raise venture capital, this sector of the pharmaceutical industrial industry stands to benefit the most from smaller and less time consuming clinical trials. Right now, the dropout rate from Phase III—the last barrier to market is about 50 percent, and almost half the total costs will be incurred by then. It has become common practice for small biotech companies to endure a forced exit, a sale to a larger company, because venture capitalists are resistant to having their stock diluted in yet another funding round when the product may never get approved. The offer from a larger company will buy them out and take the risk is very attractive.
It sounds great to just make it possible to let small drug companies break into the big drug market, as Airbnb and Uber have done with hotels and cabs, but it's not that easy.
However, it is in the case of a
Caveat:
And that caveat is biologics. Just like with pills, biologics such as vaccines need safety data established, perhaps even more so because of the chance of severe immune system reactions to the foreign proteins that are found in biological drugs. But unlike small molecule drugs, biologics like vaccines usually provide indirect evidence of efficacy even in safety trials because an immune response will normally be observed.
The presence of a strong immune response is a good, but not perfect, indicator of efficacy of a vaccine.
So conditional approval for vaccines and other biologics that generate surrogate markers of efficacy is a feasible idea, especially in the case of unmet medical needs.
Yet those instances are limited. Outside that, a new FDA head won't realistically be able to streamline the FDA drug approval process by approving new drugs with little or no efficacy data.
But what a new FDA head can do is make it easier for
Exceptions:
When there is a substantial change in the risk-benefit ratio of an experimental drug, rules need to reflect this. For example, some cancers (pancreatic, lung, colon) are notoriously difficult to treat. When the alternative is a certain death, then a much higher risk becomes acceptable.
It could be argued that drugs for previously untreatable, fatal conditions should be given a status enabling them to be conditionally approved for dying patients.
Conditional approval would be contingent upon continuing studies of the drug.
Suggestions:
- There is a perennial backlog pending reviews, both for new and generic drugs. Rather than blowing up the system, this could be alleviated by hiring more FDA scientists to evaluate new drugs.
- The FDA continues to require more and more safety data, which is shown by a surrogate marker —the number of principal investigators involved in a given trial, by year. The most striking aspect of the graph is the three-fold increase investigators participated in trials. (2)
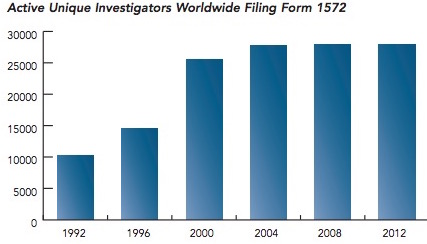
The number of principal investigators per FDA trial between 1992 and 2002. Source: Tufts Center for the Study of Drug Development analysis of the FDA’s Bioresearch Monitoring Information System
Are drugs three-times safer than in 1992? Certainly not, so it is not unreasonable to wonder why so much extra manpower (and, thus time) is now built into the system, but offers little benefit.
Conclusion:
Although it may make appear that approval of drugs based on safety data alone would be one way to cut through red tape and see if the drug really works, it would do just the opposite.
The lack of dosage guidance, and (especially) the absence of any real-world way to determine collect and analyze efficacy data would bring the process to a screeching halt.
Notes:
(1) On average, patients try 2-3 antidepressants before finding the one that is best for them. This varies greatly from one individual to another.
(2) Form 1572 is a "Statement of Investigator." All investigators must complete it before they can participate.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


