If you watched professional wrestling in the 1960s it would be difficult not to remember William "Haystacks" Calhoun. Calhoun, always a crowd favorite, had a signature move called "The Big Splash," where he bounced off the ropes and the hurled himself on top of his opponent, who was lying helplessly in the middle of the ring. This could not have been a pleasant experience for the other wrestler; Calhoun, who died in 1989 at age 55, was 6'4" and weighed 601 pounds (1). The term "Big Splash" was not an exaggeration.
On the other end of the size spectrum, is another hugely popular celebrity of a different kind, Peter Dinklage, who steals the show as Tyrion Lannister in Game of Thrones. Dinklage, who is 4'5" tall, weighs 110 pounds.

Would Haystacks Calhoun (L) and Peter Dinklage (R) both take two tablets of aspirin? Photos: Facebook, Wikimedia, Pxhere
Now imagine that both Dinklage and Calhoun have headaches and need aspirin. The recommended adult dose for Bayer Aspirin is two 325 mg tablets every four hours. Will this dose be suitable for both men? A website called Omni Calculator makes it possible to estimate the volume of blood in a person based on gender, height, and weight.
Plug in Haystacks and you'll see that he had 12,019 mL (12.7 quarts) of blood in his body. Do the same for Dinklage, and the number is quite different - 3,105 mL (3.3 quarts). All things being equal, the concentration of aspirin in Dinklage's blood would be four-times that in Calhoun's from the same two aspirin tablets. Is Dinklage taking too much aspirin or is Calhoun taking too little? It is impossible to say, but it is all but certain that two aspirin tablets are going to elicit very different physiological responses in each man.
While the comparison may seem silly, in today's anti-opioid climate a "one-size-fits-all" mindset has become the foundation of government-dictated medicine. And it's very bad medicine. The deeply flawed policies that are being enacted as law all over the country are based on the "one-size-fits-none" concept of morphine milligram equivalents (MME) - the maximum amount of an opioid medication than is permitted per patient per day. While MME values are touted as useful predictors of the total "opioid load" that a patient can receive, they are nothing of the sort. And MME-based policies don't just fail because of differences in the size of patients; they fail for multiple reasons.
Flawed science yields meaningless results
Below is a chart published by the CDC, a "guide" (2) for physicians who prescribe pain drugs. Morphine is normalized to 1.0 and the conversion factor reflects the relative potency of other opioid drugs. So, if the daily MME - the maximum dose of drug allowed - is 90 mg (3) then a patient may receive no more than 90 mg of morphine, 90 mg of hydrocodone, 60 mg of oxycodone, or 30 mg of oxymorphone per day. Although the conversion table seems to be straightforward enough, it is based on an assumption that all opioids behave similarly in the body. But this assumption could not be less accurate. Once we see the profound differences in the properties of the drugs and the difference between individuals who take them it becomes clear that not only is the CDC chart flawed, but the MME is little more than a random number.
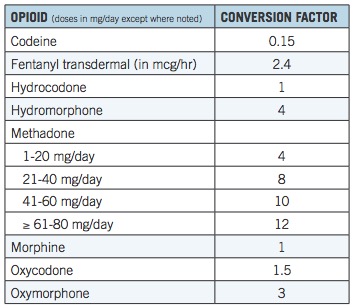
Table 1. MME equivalents. Source: CDC
Not all opioids are created equal, especially in the body
Anyone with even a passing knowledge of pharmacology would immediately be skeptical of data in the chart. Let's take, for example, the two drugs at the bottom. Although Table 1 tells us that oxymorphone is twice as "strong" as oxycodone it does not take into account a number of critical properties that paint a more complete picture of the fate of the drug once swallowed. In other words, there is no information about pharmacokinetics - the effect of the body on the drug.
Bioavailability
One of the many pharmacokinetic properties required to establish how a drug will fare within the body is called bioavailability - a critical determinant for whether a drug will be effective if taken orally, and if so, what the effective dose will be. Bioavailability is a measure of the how well a pill will be absorbed in the gut and subsequently enter the bloodstream. For example, if a drug has a bioavailability of 100% then all of that drug that was swallowed will end up in the blood (4).
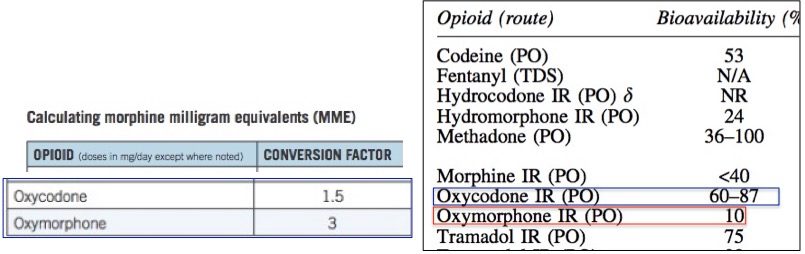
Figure 1. (Left) The MME conversion factor used by the CDC for oxycodone and oxymorphone. Values are normalized so that morphine is 1.0. According to the table, a dose of oxymorphone is "worth" three times more than that of morphine and two-times that of oxycodone. PO means orally. IR means immediate release. Source: CDC. (Right). The bioavailability of common opioid drugs. Note that oxycodone is 6-8.7 times more bioavailable than oxymorphone. Source: Pruskowski, et.al., Opioid Pharmacokinetics #307. Journal of Palliative Medicine, 19(6), 668–670(2016). doi:10.1089/jpm.2016.0024
Conversely, when a drug has poor bioavailability, for example, 10%, then most of the drug will either pass through the intestinal tract unchanged or be absorbed but then rapidly metabolized. Furthermore, drugs with low bioavailability have a greater variation from one individual to the next, making the pharmacology of a drug like oxymorphone even less predictable. The difference in bioavailability between oxycodone and oxymorphone is stark (Figure 1) even though chemically they differ only by a single carbon (Figure 2).
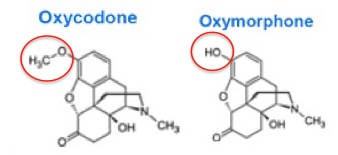
Figure 2. Oxycodone and Oxymorphone - Chemically, nearly identical. Pharmacologically, very different. The red circles indicate the only structural difference between the two - one methyl group.
Half-life and metabolism
Although critical, bioavailability is far from the only measure of an oral drug's effect on people or animals. Table 2 shows three of the most common pharmacokinetic properties of oxycodone and oxymorphone. Although poor bioavailability - one of the most daunting obstacles on the long and difficult trip from the lab to the pharmacy - has led to the demise of countless experimental drugs in clinical trials there are plenty of other obstacles. Two of these are the crucial parameters half-life and metabolism.
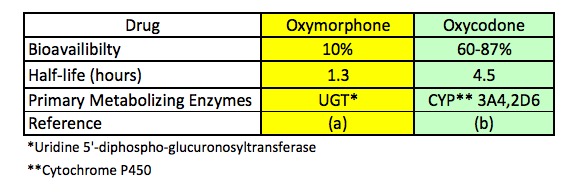
Table 2. Three pharmacokinetic parameters of oxymorphone (right) and oxycodone (left). Source: Ref. (a). Ref. (b).
Table 2 clearly shows that oxycodone and oxymorphone, although putatively similar, behave very differently in humans. Oxymorphone, even though it is twice as potent as oxycodone (Table 1), is metabolized much faster; its half-life (5) is 1.3 hours, while oxycodone, although less potent, stays in the blood much longer (its half-life is 4.5 hours). Additionally, there are differences which liver enzymes carry out the metabolism. Oxymorphone is primarily metabolized by a liver enzyme called uridine 5'-diphospho-glucurosyltranferase (UGT) while oxycodone is primarily metabolized by two different cytochrome P450 enzymes called 3A4 and 2D6. The difference in metabolizing enzymes itself is a substantial concern when comparing two different drugs, but it becomes even more so when other drugs are part of the picture.
The only certainty is uncertainty
So, which drug is better for a pain patient? Do the MME values really reflect the drugs' relative ability to relieve pain? Do half-life and bioavailability matter? Does the fact that different types of enzymes are involved in metabolism make a difference? The answer to all of these questions is "who knows?" All we are told is that patients cannot be prescribed more than 60 mg of oxycodone or 30 mg of oxymorphone per day.
Bear in mind that we have examined only two of the eight drugs on the CDC chart and only three of many pharmacokinetic properties of each drug. When other drugs and other pharmacokinetic properties are added to the mix It becomes patently obvious that the simple CDC chart provides us with numbers that are probably more artifactual than real. The chart tells us little about which opioid drug might the best for a single person or its optimal dose, let alone a diverse population. More on that below.
Polypharmacy: Drug-drug interactions
The primary site of drug metabolism is the liver. Within the liver, there are different families of metabolizing enzymes. By far, the most important class in humans is the CYP450 family (6) of enzymes. There are about 60 members in this class and these account for about 75% of the metabolic processes that occur within our bodies. Two of the most important subtypes (also called isozymes) of this family are called CYP3A4 and CYP2D6. The two are responsible for metabolizing many common drugs, especially opioids.
Table 2 shows that even two very similar drugs will behave differently when taken alone, but when other common drugs are taken, the variability of opioid blood levels becomes even greater; opioid-metabolizing enzymes can be profoundly affected by other drugs. As a result, these enzymes can be inhibited (less metabolism) or induced (more metabolism), each of which will have a pronounced effect on the blood levels of an opioid. Figure 3 provides a summary of common drugs that alter opioid blood levels (7). 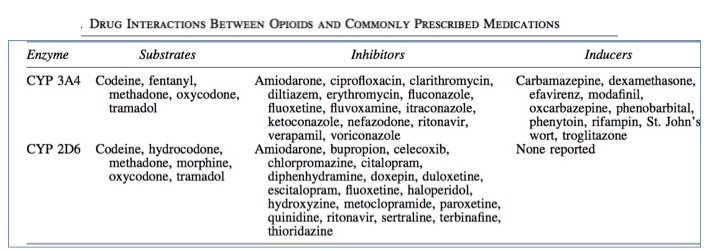
Figure 3. Drug-drug interactions of selected opioids with some common drugs. Source: J. Pruskowski and R. Arnold, Opioid Pharmacokinetics #307, Journal of Palliative Medicine Vol. 19, No. 6 (2016). doi.org/10.1089/jpm.2016.0024
Some highlights from Figure 3 include:
- Of the six opioids listed, five of them are metabolized by both 3A4 and 2D6. Morphine is not metabolized by 3A4.
- The 3A4 isozyme is inhibited by many drugs, including antibiotics, antifungals, antidepressants. blood pressure drugs, and HIV antivirals. The presence of any of these drugs will either result in higher than expected opioid blood levels, an increased half-life of the opioid, or both. Inhibitors of 3A4 can increase blood levels of the opioid (except for morphine - it is not metabolized by 3A4), sometimes causing dangerously elevated levels of the opioid.
- The 3A4 isozyme is also induced by other common drugs, such as anti-inflammatory steroids, HIV antivirals, anti-seizure drugs, and a tuberculosis drug. The presence of any of these drugs will either result in lower than expected opioid blood levels, a decreased half-life, or both. This can result in opioid levels that are inadequate for pain relief.
- The 2D6 isozyme is inhibited by many of the same drugs that also inhibit 3A4 but also others, including those for allergies, malaria, and schizophrenia.
- But the 2D6 isozyme is not induced by any of these commonly used drugs.
The clinical impact of drugs that inhibit 3A4 becomes apparent in a case study of a cancer patient who was being treated with fentanyl patches for pain. While hospitalized, the patient suffered a near-fatal overdose of fentanyl at a dose that would not normally be problematic; he was also taking an antibiotic to treat a bacterial infection (erythromycin) and an anti-fungal drug (itraconazole) to treat the fungal infection caused by the erythromycin. Both drugs are potent inhibitors of 3A4, which led to elevated levels of fentanyl, resulting in impaired breathing. Once the 50 microgram patch was replaced by a 25 microgram patch the breathing difficulties resolved.
Other studies (8,9) have demonstrated that concomitant use of an anti-fungal drug increased the blood levels of oxycodone four-fold, while a drug for tuberculosis greatly decreased oxycodone levels.
Genetics: Abundant differences in human metabolism of opioids
Perhaps the most important factor in determining the optimal dose of an opioid is the profound difference in the genetic makeup of individuals; some people may metabolize opioids 100-times that of others simply because of the impact of genetics on CYP function. Table 3 shows the range of variation of the rate of metabolism of four CYP enzymes.
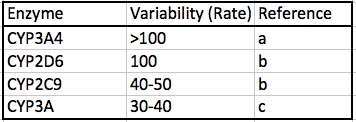
Table 3. The range of individual variation of the rates of metabolism for four CYP isozymes. Note that the first two, 3A4 and 2D6, are the primary opioid metabolizing enzymes, as shown in Table 2.
References:
(a) Pharmacogenomics of Cytochrome P450 3A4: Recent Progress Toward the “Missing Heritability” Problem. K. Kleinand Ulrich and M. Zanger Front Genet. 2013; 4: 12. (2013) doi: 10.3389/fgene.2013.00012
(b) Hart SN, Wang S, Nakamoto K, Wesselman C, Li Y, and Zhong XB (2008) Genetic polymorphisms in cytochrome P450 oxidoreductase influence microsomal P450-catalyzed drug metabolism. Pharmacogenet Genomics 18:11-24.
(c) Westlind A, Lofberg L, Tindberg N, Andersson TB, and Ingelman-Sundberg M (1999) Interindividual differences in hepatic expression of CYP3A4: relationship to genetic polymorphism in the 5'- upstream regulatory region. Biochem Biophys Res Commun 259:201-205.
Given the wide range of CYP activity variability from individual to individual, it should come as no surprise that this difference profoundly affects opioid users, especially since CYP3A4 and CP2D6 are responsible for much of metabolism of these drugs. It is this genetic variability that is responsible for both poor metabolizers and rapid metabolizers of opioids.
For example, if the innate rate of metabolism by CYP34A of Patient A is 100-fold greater than that of Patient B, it will be impossible to define a standard dose, such as in Table 1, which would be therapeutically appropriate for either A, B, or both. Patient A will appear to have developed a tolerance for the opioid while Patient B will be far more sensitive to the drugs. Thus it is almost a given that any standardized dose will be too low for Patients like A and too high for Patients like B.
Problems and more problems:
This article is a cursory summary, not a comprehensive review of all the factors that combine to make opioid dosing anything but simple. Yet, the following are obvious:
- Some opioid drugs will be absorbed and pass to the bloodstream very well and some will do so very poorly.
- Even opioids that appear to be structurally and functionally similar will be metabolized at very different rates.
- Other drugs can drastically alter the physiological response of a pain patient to a given opioid; the second drug may increase a person's response to the opioid or it may decrease it.
- Even under ideal conditions - two people taking the same opioid drug at the same dose, at the same interval, and taking no other drug - huge variations of innate metabolism from one individual to another will necessarily result in a wide range in clinical response to that drug.
Conclusion
The CDC MME chart, in fact, the entire concept of morphine milligram equivalents may be convenient for bureaucrats but because of differences in the absorption of different drugs into the bloodstream, half-life of different drugs, the impact of one or more other drugs on opioid levels, and large differences of the rate of metabolism caused by genetic factors, is not only devoid of scientific utility, but actually causes far more harm than help by creating "guidelines" that are based upon a false premise. When a policy is based on deeply flawed science, the policy itself will automatically be fatally flawed. It cannot be any other way.
Policy Recommendations:
- The 2016 CDC Guideline for Prescribing Opioids for Chronic Pain and all its findings should be immediately withdrawn.
- A new set of guidelines must not be unilaterally formed by a single agency, especially the CDC, which lacks the chemical and pharmacological insight and knowledge to formulate a policy regarding drug use.
- The FDA has a far greater knowledge base about drugs and pharmacology and should form a task force composed of in- and outside pharmacologists, chemists, pain management physicians, neurologists, primary care physicians, surgeons, and pain patients.
- The task force should formulate a science- and medicine-based set of recommendations that are focused on treating and preventing pain, not stopping addiction - an endeavor that been shown time and time again to make matters only worse.
- The task force should submit its plan to the Commissioner of the FDA for review.
- Personalized medicine is the hallmark of the 21st Century. While it is now commonplace in oncology and cardiovascular disease personalized medicine lags behind in pain therapy. Technology now exists to quickly identify the genes that make fast- and slow-metabolizers of opioid drugs and, much like oncology, use a genetic profile to identify a safe and effective dose for individuals who are at risk of an overdose and those who need higher doses or medication simply because their personal metabolism requires them. Cheek swab tests that give individual CYP profiles are available.
- Medication for pain is no less important for that of cancer or heart disease. Funding research for profiling individuals and tailoring treatments for pain patients will go a long way in clarifying the fuzzy boundary that exists between use and abuse of powerful narcotics.
- Patients should not be viewed as potential addicts nor should pain specialists should be viewed as owners of pill mills.
- Physicians must be given the required independence and tools necessary to empower them to treat individual patients as they see fit without undue interference from punitive legislation and overzealous law enforcement.
- There are two faces of the opioid crisis - overdose deaths and pain remediation. These are very different and must not be conflated.
NOTES:
(1) Calhoun's weight increased as he aged. He was said to weigh more than 700 pounds late in his career.
(2) The "CDC Guideline for Prescribing Opioids for Chronic Pain" was nothing of the sort. In reality, it was the basis for the widespread legislation that codified the maximum quantity of prescription drugs that was allowable per patient. This is unprecedented in US history.
(3) For some unknown reason 90 MME was chosen as the magic maximum. This number will be too high for some and too low for others. No simple number can define the "proper" dose of any drug, nor should it.
(4) This is an oversimplified explanation of bioavailability. In reality, bioavailability is a ratio of the concentration of the drug the blood when given orally compared to that when given intravenously.
(5) The half-life of a drug, which is abbreviated as t1/2 is the amount of time it takes for half of the drug to be metabolized. For example, if a drug has a half-life of one hour, 50% of the drug will remain after an hour. After two hours, 25% will remain. The drug is generally considered to be "gone" after four half-lives; only 6.25% of it will remain.
(6) CYP450 is an acronym for CY = cytochrome, P= pigment, and 450 refers to the wavelength of light (in nanometers) that it absorbs when exposed to carbon monoxide. The numbers and letters that follow classify the CYPs into different families, which are also called isozymes.
(7) These lists are incomplete. Comprehensive lists of drugs that inhibit or induce CYPs are considerably longer.
(8) Hagelberg NM, Nieminin TH, Saari TI, et al. Voriconazole drastically increases exposure to oral oxycodone. Eur J Clin Pharmacol. 2009;65(3):263-271.
(9) Nieminen TH, Hagelberg NM, Saari TI, et al. Rifampin greatly reduces the plasma concentrations of intravenous and oral oxycodone. Anesthesiology. 2009;110(6):1371-1378.,

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.



{kind=link}