
# Reprinted with permission. This article originally appeared in Current Medical Research and Opinion.
ABSTRACT
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) created an abbreviated licensure pathway in the United States that allows for the development and approval of biologic products shown to be biosimilar to or interchangeable with a US Food and Drug Administration (FDA)-licensed reference product. FDA released the draft guidance for industry on Demonstrating Interchangeability with a Reference Product (hereafter referred to as the Draft Interchangeability Guidance) in January 2017. Despite FDA’s efforts, there continues to be a great deal of confusion and misinformation surrounding the topic of interchangeability. Here we discuss interchangeability, as well as substitution of biological products, with a focus on the US. Additionally, the separate topic of physician-mediated switching is covered and distinguished from interchangeability and substitution.
Interchangeability
The term interchangeability has different meanings in different parts of the world, and substitution practices also vary by jurisdiction. Here we focus on the developing situation in the United States. Interchangeability is statutorily defined in the BPCIA to mean that the product “may be substituted for the reference product without the intervention of the health- care providers who prescribed the reference product”. To be deemed interchangeable, a biological product must be biosimilar to an FDA-approved reference product and meet additional standards for interchangeability set forth by the BPCIA. Specifically, in order for FDA to deem a biological product interchangeable with a reference product there must be sufficient information to show that:
the proposed interchangeable product can be expected to produce the same clinical result as the reference product in any given patient; and the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch (for a biological product that is administered more than once to an individual).
The statutory requirement that sufficient information must be provided to show that the biological product “can be expected to produce the same clinical result as the reference product in any given patient” could be conservatively interpreted to mean that, in order for the FDA to deem a proposed product interchangeable, clinical data in each indication for which the reference product is licensed must be provided in the application (or supplement). This does not seem to be the Congressional intent, however, since interpreting the statutory language in this manner would necessitate such an amount of clinical data that the pathway would be unlikely to be utilized. Extrapolation of data to conditions of use not directly studied in clinical trials is an essential concept to both biosimilarity and interchangeability that is consistent with FDA guidance and enables an abbreviated pathway.
According to the Draft Interchangeability Guidance the FDA does expect data from a clinical switching study, evaluating two or more alternating switches, in order to meet the statutory requirement that sufficient information must be provided to show that “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch”. A switching study intending to support a demonstration of interchangeability could be conducted as a dedicated study (separate from the comparative clinical study demonstrating biosimilarity) or integrated with the comparative clinical study. Regardless of design, the data standards are the same. Figure 1 depicts what an integrated switching study design may look like.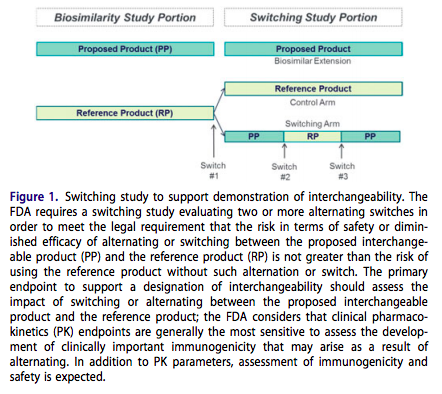
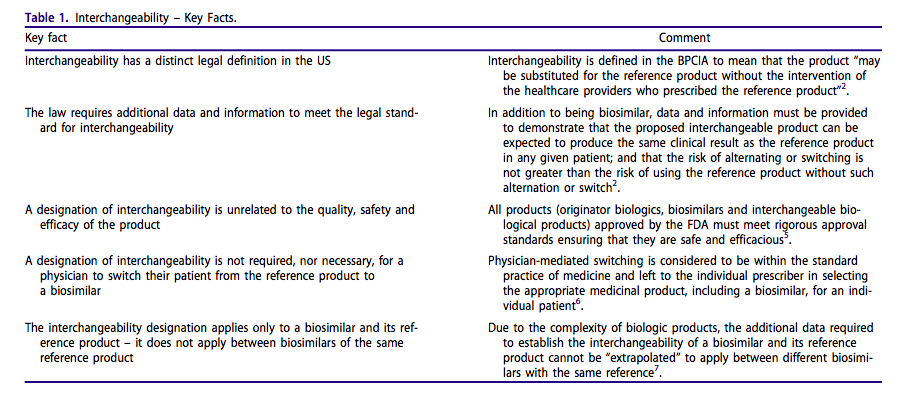
It is important to recognize that a designation of interchangeability does not relate to product quality or degree of similarity to the reference product. Rather, this designation signifies that the interchangeable biological product sponsor has provided the FDA with the additional data and information necessary to meet the statutory standard for substitution. The FDA has stated that “from a quality perspective, a product that is first approved as a biosimilar is not expected to be changed in some manner to ‘become’ an interchangeable product”3. Ultimately, it is at the biosimilar manufacturer’s discretion whether or not to seek an interchangeability designation for a particular biosimilar. Table 1 Summarizes Key Facts about interchangeability in the US.
Substitution
In the US, although Federal law gives the FDA the authority to license biological products as interchangeable, it is the state laws that govern the substitution of medicines. A product may be substituted at the pharmacy without consulting the prescribing physician where there has been a scientific determination of licensure as an interchangeable biological product by FDA, and where state law permits substitution of biological products.
Figure 2. Single cross-over data to support physician-mediated switching of non-treatment-naïve patients. An interchangeability switching study (Figure 1) is not required to support physician decisions to change a patient’s course of therapy. Comparative single transition data, as depicted here, may be required to support a demonstration of biosimilarity where it may be considered a clinically relevant scenario for non-treatment-naïve patients to transition to a biosimilar product from the reference product.
Beginning in 2013 a coalition of stakeholders made of patient groups, provider organizations and biopharmaceutical companies have supported passage of legislation to ensure that state laws are up to date concerning substitution of interchangeable biological products.
Biosimilar prescribing and physician- mediated switching
Biosimilars, and interchangeable biologics, can be prescribed by healthcare providers (HCPs) in the same way they prescribe other medications. In contrast to products deemed interchangeable, non-interchangeable biosimilars must be specifically prescribed by HCPs and cannot be substituted at the pharmacy level. The term “physician-mediated switching” describes a prescribing decision made by a physician to change a patient’s treatment – to another course of therapy, between reference product and a biosimilar, or potentially between biosimilar products. There is no statutory standard for physician-mediated switching. Rather, such physician- directed decisions are part of usual medical practice and are independent of, and not in any way related to, substitution. A determination of interchangeability by the FDA is not required for a physician to prescribe a biosimilar to a patient who is being treated with the reference product.
Under certain circumstances the FDA may require descriptive data assessing safety and immunogenicity following a single cross-over or transition from the reference product to the proposed biosimilar to support a demonstration of biosimilarity. Where required, the single cross-over from reference product to the proposed biosimilar is included following the assessment of the primary efficacy endpoint as part of the comparative clinical trial conducted to demonstrate there are no clinically meaningful differences between the proposed biosimilar and the reference product (Figure 2). Comparative single transition data of this nature is not sufficient to meet the statutory expectations for demonstration of interchangeability. This single transition data is generally necessary to support a demonstration of biosimilarity, for example with a chronically
administered product where it may be considered a clinically relevant scenario for non-treatment-naïve subjects to transition to a biosimilar product from the reference product.
The topic of switching is of great interest to the medical community and other stakeholders. Real world data is being generated at a rapid pace, especially in Europe where biosimilars have ten-plus years of on-market usage. A systematic literature review was recently published that describes 90 studies encompassing in excess of 14,000 unique individuals and seven different molecular entities used to treat 14 diseases. The authors concluded that the risk of immunogenicity-related safety concerns or diminished efficacy is unchanged after switching.
Conclusion
A designation of interchangeability, or lack thereof, does not relate to the quality, safety or effectiveness of biosimilars or interchangeable biological products. Rather, this designation is intended to meet an additional statutory standard that would enable substitution at the pharmacy level without the intervention of the healthcare provider who prescribed the reference product. Physicians’ confidence in prescribing biosimilars to their patients, including and where appropriate to patients already receiving the reference product, should not be impacted by whether the product has been deemed interchangeable by the FDA. Whether a product is biosimilar to or interchangeable with a reference product, patients and physicians should be assured that the product has met the FDA’s rigorous approval standards for quality, safety and efficacy.
Dr. Robert Popovian completed his Doctorate in Pharmacy and Masters of Science in Pharmaceutical Economics and Policy degrees at the University of Southern California and completed a residency in Pharmacy Practice/Adult Internal Medicine and Infectious Diseases at the Los Angeles County – University of Southern California Hospital where he was the head pharmacy resident (primary area of research: gram negative bacteremia) and a fellowship in Pharmaceutical Economics and Policy at University of Southern California. He is currently Senior Director, US Government Relations at Pfizer Inc.


