Dr. Katherine Seley-Radtke, a professor of chemistry and biochemistry at the University of Maryland, Baltimore County is an expert in antiviral drug discovery, and a member of the ACSH Scientific Advisory Board. Dr. Seley-Radtke and I cautioned that an antiviral drug, not a vaccine, may prove to be the best way to get Covid-19 under control in our July 1st op-ed in the Baltimore Sun.
She generously agreed to spend some time telling us what she believes to be a path forward and also explain her group's unique strategy in designing drugs that inhibit RNA virus in general and SARS-CoV-2 in particular.
JB: Dr. Radtke, first I want to thank you for answering some of my questions about what I believe is an exciting approach that stands a good chance of providing the kind of potent drugs that may be needed to get an upper hand against a very scary infection.
KR: I'm happy to do it. People need to know what antiviral drugs are capable of. And I see no reason why the strategies that have been given us drugs that were enormously successful against other viruses won't do the same here.
JB: What is your take on the ongoing effort to find a useful COVID drug from a collection of approved drugs, most of which were not designed to treat any virus, let alone SARS-CoV-2 (coronavirus)?
KR: I think it is a huge waste of time testing most of them. Testing antiviral drugs? Yes, absolutely. Testing random drugs that aren’t direct-acting antivirals is just wasting valuable time that could be focused on finding a cure.
JB: Can you explain why even an antiviral drug that is approved for treating a certain infection will not necessarily work against a different virus, perhaps even one in the same family?
KR: Drugs bind in certain places in viral enzymes known as a binding site and when they do so effectively it can stop the virus from replicating. All binding sites are made up of various amino acids with which the drug interacts in a very specific way. The reason for this specificity is that the binding sites of related viral enzymes, in this case, viral polymerases – enzymes responsible for building the viral genome – are different from the others. So, even within the same virus family the action of one drug may differ significantly from another. In other words, what binds in one doesn’t necessarily bind in another, or may do so less effectively. This recognition between the binding site and the drug is critical for the drug’s activity.
JB: The potency of antiviral drugs is measured by a parameter called the EC50. Can you tell us how this number is determined and what it means?
KR: EC50 stands for the minimum effective concentration of a drug that leads to the maximum effect for 50% of whatever is being tested – animals, humans, or even a virus in a test tube. In the case of viruses, the EC50 of a potential drug would be the concentration of the drug required to stop 50% of the replication – something that can easily be measured. It is important to note that the more potent the drug, the lower the EC50. So, a drug with an EC50 of 0.20 micromolar against a virus is five-times more potent than a different drug with an EC50 of 1.0 micromolar (1).
JB: If all the repurposed drugs fail, or work poorly what is your best guess of the reason?
KR: Because the viral enzymes don’t recognize the drug, either because they are not antivirals or because the drug doesn’t fit well enough in the polymerase enzyme’s binding site.
JB: If you had to pick a single strategy for taming COVID what would it be and why?
KR: I think our best shot is a drug cocktail made up of one or more nucleosides and probably another class of inhibitor such as a protease, integrase, or entry inhibitor. These have all been used to battle other viral infetions. But the foundation of the cocktail will undoubtedly be a nucleoside because as a class, they have historically had the best success at treating viruses of many different viral families.
I think it is a huge waste of time testing most of [the repurposed drugs]. Testing antiviral drugs? Yes, absolutely. Testing random drugs that aren’t direct-acting antivirals is just wasting valuable time that could be focused on finding a cure.
Dr. K. Seley-Radtke, 8/13/20
JB: Can you give us an example of one or two of antiviral drugs that work this way?
KR: Yes, for example, Harvoni is a two-drug cocktail that cures hepatitis C. It contains a nucleoside inhibitor – sofosbuvir also known as Sovaldi – as its primary component. Another example is Biktarvy, a potent anti-HIV combination. Biktarvy is made up of two nucleosides – emtricitabine and tenofovir alafenamide, as well as a third drug bictegravir, which is an integrase inhibitor. There are many other nucleoside analogues that inhibit different viruses, and they all work by inhibiting one of the viral enzymes involved in that virus’s replication pathway – polymerases.
JB: Your group is working on a class of very interesting analogs of nucleoside polymerase inhibitors called fleximers. What is a fleximer?
KR: A fleximer is a type of nucleoside analogue that I invented about 20 years ago. Nucleosides are made up of two basic key pieces – a sugar and a heterocyclic base. The key structural feature of a fleximer involves the heterocyclic base, which in many nucleosides is a fused bicyclic, two-ring compound, known as a purine. In the fleximer, the purine base has been split apart into its two components but stuck together by a single carbon-carbon bond. This gives them the ability to rotate and reposition in the enzyme’s binding site (Figure 1).
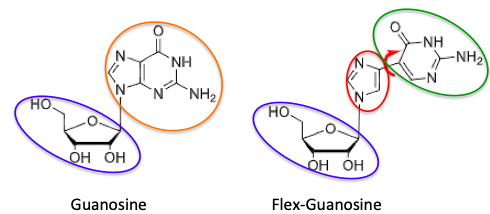
Figure 1. The structures of guanosine (left) and flex-guanosine (right). The sugar (blue) fragments are the same in both. But in flex-guanosine, the bicyclic purine ring (orange) is split into its components (red and green) and these are reconnected with a single bond. The red arrow shows the freedom of rotation gained from this transformation.
Because of this flexibility, it gives them some very strategic properties – the ability to bind with better amino acid residues than the normal nucleoside can, which in turn, can lead to greatly increased activity, as well as the ability to overcome resistance mechanisms - this is when the virus develops mutations in the binding site so that the drug no longer binds. The fleximers can overcome that by binding with different amino acids in order to retain their activity. In addition, the ability to change their conformation also can lead to inhibition of enzymes that the parent normal nucleoside has no effect on.
(Before we proceed the following table will give you an approximate idea of a range of EC50 values and how likely they are to be potent enough to become a drug. These are not literature values; they represent my experience in drug discovery. Other chemists might differ with my approximate assignments. Too bad. You wanna write this? Didn't think so.)

JB: Can you give us an idea of the potency of fleximers compared to other antiviral drugs?
A: Some of our fleximers that were based on acyclovir, an FDA-approved drug for treating herpes virus, have shown very potent activity against viruses like Dengue, Yellow Fever, Zika, Ebola and Marburg viruses, as well as coronaviruses such as SARS, MERS and human coronaviruses. The best activity we have seen is 57 nanomolar against Dengue, followed closely by Yellow Fever (0.3 micromolar), and Ebola at 1.49 micromolar, and the rest in single-digit micromolar. This is quite significant.
JB: Fleximers were originally investigated as herpes drugs, but they didn't work, but they inhibit SARS-2. Is there an explanation for this? More importantly, are any of them being evaluated as potential COVID drugs?
KR: These particular analogues were originally designed to inhibit herpes virus; they are fleximer analogues of the drug acyclovir. However they showed no activity against herpes, yet very potently inhibit all of those other viruses. We're not sure why.
And yes, they are now being tested against SARS-CoV-2. They should be in infected animals in the next few weeks. It is likely that the reason they exhibit broad spectrum activity across several viral families is their ability to adapt to the different enzymes better than a rigid nucleoside can. We like to call them “molecular chameleons” because like a chameleon that has to change colors to adapt to its environment, so as not to be eaten, the fleximers can adapt to their environment to stay active.
JB: How have you adapted the Ebola fleximer strategy to try to make better SARS-2 drugs?
KR: We will see!! The compounds are currently being tested against SARS-2. They are the same ones that worked against ebola, SARS and MERS etc. Based on their activity we may change their structures or maybe change the pro-drug portion of the molecule (which is the same used by Gilead to make remedesivir) to see if that increases the activity, but we are waiting on testing results first.
JB: Can you give us a simple explanation of the differences between remdesivir and your best candidates?
KR: Most importantly, our compounds are orally bioavailable – in other words, they can be administered orally (remdesivir is IV only. This is important because that is one of the huge limitations of remdesivir. Another is the synthesis – remdesivir is a bitch to make – our compounds can be made quickly and cheaply. And soon we will know if they are more active than remdesivir. Importantly, however, one issue that is likely to arise is the development of resistance. We are already seeing numerous strains out there of CoV-2, so the ability to overcome resistance will be important and the fleximers can do this, remdesivir cannot.
JB: How does this mess play out? What's your best guess?
KR: Hopefully in the coming months we will have several small molecules either repurposed or new drugs that can be used in combination to treat this pandemic. More importantly, our goal is not just to find a cure for SARS-CoV-2, but to develop a broad spectrum inhibitor that will be ready to use when the next outbreak of a new or re-emerging virus makes its appearance. There are hundreds, if not thousands of zoonotic diseases out there just waiting to jump from animals to humans, and we have to be ready. We should never find ourselves this unprepared again!
NOTE:
(1) One of the problems with some of the repurposed drugs is their weak potency; EC50 values are in the 1 millimolar (or higher) range. A 5 millimolar drug is probably going need to be given in a high dose, which usually means limited efficacy and probably side effects. All things equal, potent is always better.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.