Pre-exposure prophylaxis (PrEP) is one of several historic breakthroughs in the four-decade battle against HIV/AIDS. There are several PrEP drug combinations, all very effective in preventing people from contracting HIV from HIV+ individuals, even in the absence of protection, such as condoms. These (and similar) drugs also treat people who are HIV+ from becoming ill by keeping the viral load below measurable levels, at which point they cannot transmit the virus to others.
Pfizer recently conducted a conceptually similar clinical trial called Post-Exposure Prophylaxis (PEP) to determine whether Paxlovid, the most effective Covid antiviral drug, could act to prevent the infection of uninfected members of households where someone had confirmed Covid. The answer? No, something the company announced in a recent press conference and was widely reported as bad news.
I must respectfully disagree. It is not bad news. The very last thing we need is to have people who don't need Paxlovid taking it "just in case." Why? Adaptive resistance. Viruses are masters of morphing in response to a drug challenge (especially a single drug) (1) to create mutant strains that are no longer as susceptible to that drug, possibly even unaffected by it. The more the drug is used, the greater the chance of developing these strains, so given that Paxlovid is by any measure an essential drug, it needs to be used only when necessary to slow the inevitable resistance.
There are a number of different ways that viruses can create mutant strains that are less susceptible to a drug, the most common being by changing the shape of the area where the inhibitor (drug) binds, resulting in a poorer fit. Let's look at a simple example from the world of hepatitis C, specifically, the HCV protease inhibitor telaprevir.
A Single Amino Acid Mutation Screws Up Telaprevir
Although it is no longer used (2), telaprevir (Brand name Incivek) represented a historic breakthrough therapy for hepatitis C because it (3) was the first-ever direct-acting antiviral drug that was approved to cure hepatitis C (HCV). Telaprevir's mechanism of action was the inhibition of HCV protease; the research used to create it was an extension of HIV/AIDS research (4).
Although resistance is not why the drug ultimately failed, a study published in the journal Antimicrobial Agents and Chemotherapy shows that a single amino acid mutation in HCV protease, which was driven by exposure to the drug (5) resulted in a decrease in the potency of the drug against the new strain – exactly what you'd expect. This is a quintessential example of selection pressure (aka evolution) in viruses.
Don't let the title of the paper – "Characterization of V36C, a Novel Amino Acid Substitution Conferring Hepatitis C Virus (HCV) Resistance to Telaprevir, a Potent Peptidomimetic Inhibitor of HCV Protease" intimidate you. In English, this means a mutant strain of HCV in which the amino valine at position 36 of the protease enzyme was replaced by cysteine, another of the 20 amino acids that make up proteins. (I discussed the mutation of amino acids in Covid in a recent article.) The result? Telaprevir lost some of its ability to inhibit the strain with the V36C mutation compared to the wild-type protease. You should be able to see this, hopefully without wanting to hang yourself, in Figure 1.

Source: Antimicrobial Agents and Chemotherapy, Vol. 54, No. 6, DOI:https://doi.org/10.1128/AAC.01796-09
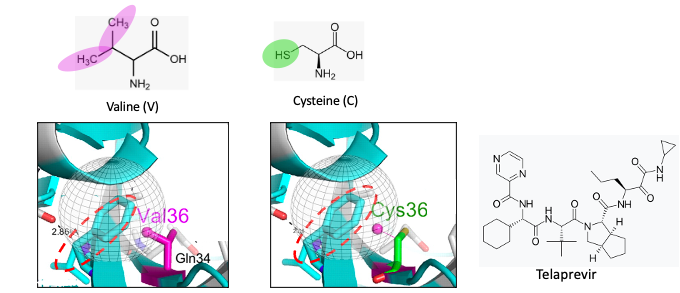
Figure 1. Top/left: The chemical structure of valine. The magenta ovals show the isopropyl group found in the amino acid valine. Top/right: The chemical structure of cysteine. The green circle shows the thiol group found in cysteine. Bottom: The gray globe indicates the hydrophobic binding pocket in the protease enzyme where telaprevir binds. To keep this as simple as possible, hydrophobic (water-hating, oil-loving) pockets in proteins bind to hydrophobic fragments of molecules (like dissolves like), typically those with a preponderance of carbon atoms rather than oxygen or nitrogen. Valine, a hydrophobic amino acid is one of nine hydrophobic amino acids that would be expected to be part of a hydrophobic binding pocket.
(Bottom/left): Molecular model showing a whole bunch of stuff (hang in there). The light blue pasta-looking thing (called a ribbon) is a tiny fragment of HCV protease. The red hatched circle indicates a small fragment of telaprevir, which binds to that particular region of the protease. It is (unfortunately) also light blue. Perhaps they were having a sale on the color that day. The take-home message is that when valine is replaced by cysteine, the hydrophobic pocket becomes less so. Telaprevir now binds less tightly to the protease (it is a poorer inhibitor).
English translation: A single mutation in the structure at the telaprevir binding site on the protease weakens the binding between telaprevir and the protease, making telaprevir a less potent inhibitor. In other words, the drug will not work as well.
What Does "Not Work as Well" Mean?
To put this in perspective, the molecular weight of HCV protease is 22,700 daltons (atomic mass units). Valine is 117 and cysteine is 121. If you think about it, it's quite amazing that a switch of two amino acids with a net difference of 4 daltons in a protein with a mass of nearly 23K daltons makes such a difference in the properties of the mutant enzyme that a previously-potent drug is now less so. How much less?
The authors provide two measurements of V36C, both valid:
- In an enzyme-based (binding) assay, telaprevir's binding to the protease is 4-times weaker (its IC50 is 4X higher) vs. the V36C mutant.
- In a cell-based assay, that difference is 10-fold.
Relevance to Paxlovid (Nirmatrelvir)
Although no variants containing mutated regions where nirmatrelvir (6) binds have been isolated so far, it is inevitable that such mutations will occur. How fast? This is unknown. But what is known is that the more an enzyme "sees" an inhibitor the more likely it is to develop resistance to that inhibitor. This is why it is essential that Paxlovid access be limited to people who are actually sick, not people who are healthy but afraid of becoming sick.
The Clock is Ticking - Several Clocks, Actually
Although (for now) the death toll from the newer Omicron subvariants remains relatively low, there are some troubling signs on the horizon.
- Paxlovid will eventually become less effective.
- At this time there is nothing in development to replace it.
- As Omicron continues to mutate there is evidence that it is becoming more able to evade both vaccine- and infection-acquired immunity. It is now routine for people to catch Covid twice, even when fully vaccinated. And it won't necessarily be mild disease either.
- Although newer vaccines are in the pipeline they won't be here anytime soon. By the time they are available, they could already be obsolete.
At this time, the only thing that is keeping Covid from once again becoming a very scary disease, especially to the elderly and unhealthy, is Paxlovid. I don't even want to think about what life would be without the one magic bullet we have to prevent a possible return to the early days of the pandemic.
It is fortunate that the Pfizer trial failed. The very last thing we need is careless use of this drug.
NOTES:
(1) It is much more difficult for viruses to mutate when they are "hit" by two different drugs that operate by different mechanisms.
(2) Telaprevir and its "twin" from Merck, boceprevir became quickly obsolete once much better drugs, like Sovaldi, became available.
(3) Although neither telaprevir nor boceprevir was a financial success (quite the opposite, really) they were nonetheless milestone drugs. Hepatitis C was discovered in 1989. It took 22 years for these two – the first antiviral drugs to cure the infection – to be discovered and developed.
(4) Invirase (1995) was the first HIV protease inhibitor and the first drug that began to lower the death rate from AIDS. Scientists used the same strategy to discover telaprevir and boceprevir.
(5) There is WAY too much in the Antimicrobial Agents and Chemotherapy to discuss here the V36C mutation was found (obviously) in drug-treated patients but also in at least one drug-naive patient. This speaks volumes about viral mutation.
(6) Paxlovid is a combination of nirmatrelvir, the antiviral drug, and ritonavir, a "booster" that reduces the metabolism of nirmatrelvir.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


