
Sickle cell disease, SCD, is a genetic mutation in hemoglobin that alters the shape of blood cells, making them sickle-shaped rather than round. While it confers protection against malaria, more importantly, the abnormally shaped cells obstruct blood flow, causing significant pain and organ damage. SCD results in increased hospitalizations and mortality and generally hurts the quality of life. There are several medications available to treat SCD, but none are curative. Stem cell transplantation can be curative, although it carries a significant risk of rejection. Gene therapy using CRISPR-Cas9 technology to replace abnormal hemoglobin is tantalizingly close and is now being clinically trialed.
One of the difficulties in managing Alzheimer’s Disease is that, as with SCD, there is, up to now, no cure, and the medical treatments are far from restorative. The recent approvals for a variety of medications for Alzheimer’s with little efficacy but the enthusiastic desire on the part of patients suggests that in this situation, hope triumphs over scientific objectivity. But with the past week’s FDA review of a gene therapy for SCD, the situation may be dramatically changed.
A brief detour into Exa-cel, a gene therapy
The treatment involves a CRISPR/Cas9 edit of a patient’s blood stem cells to produce an earlier form of hemoglobin, fetal hemoglobin, that is a far better oxygen carrier than the hemoglobin created by sickle cells. The increased oxygen-carrying capacity results in far fewer vaso-occlusive crises (VOC), moments when abnormal cells close off tiny blood vessels, resulting in severe pain, kidney failure, and stroke, among other manifestations. Stopping VOCs would go a long way to restoring the 20 to 30 years of life expectancy lost with sickle cell disease.
The stem cells are harvested from patients and treated with this CRISPR technology. In the meantime, patients undergo chemotherapy, which is not without significant side effects, to remove the diseased stem cells from their bone marrow. At this point, the CRISPR-treated stem cells are returned to patients intravenously.
The issue before the FDA panel was not efficacy; 29 of the 30 patients treated with exa-cel went from 4 VOC events annually to none over an average follow-up of 22 months. Nor was it safety, although the chemotherapy necessary results in significant infertility. [1] The concern was that CRISPR/Cas9, for all of its specificity, might, in fact, make alterations in other genes, “off-target effects.” You can get a much better understanding of the issues and discussion in an excellent report by Stat.
“I was in so much pain that I couldn’t lift my legs to sit on a bedpan. I couldn’t lift a fork or wash my face. I had lost control of my body. … [Approval of exa-cel will] change the lives of people suffering from this disorder. They will feel hope again, just like I did.”
– Victoria Gray, the first person treated with exa-cel
With a gene therapy for SCD in the wings, researchers sought to uncover what tradeoffs between risk and benefit patients and families with SCD would make.
What is hope worth to patients?
Participants were offered two “gene” therapies, which differed concerning their benefits, becoming asymptomatic vs. greater life expectancy, and their risks, including death and a greater lifetime risk of cancer. In each choice, participants could also choose the ‘No Gene Therapy’ option – no enhanced risk, no increase in life expectancy, and a continuation of the symptoms they had experienced over the last year.
Participants were characterized based on their symptoms over the preceding year. All participants answered questions based on their current symptoms and a worsening of their condition from increasing pain, hospitalizations, or a stroke.
- 174 adult patients and 109 parents participated in the survey.
- The adult patients’ average age was 34; 73% were female, and 97% Black. They averaged 3 VOC crises requiring medical care in the prior year; 58% had chronic pain lasting for six months or more, and 4% had experienced a stroke.
- The average age of the parent of a child with sickle cell disease was 40, again predominantly female and Black. The average age of their child was 9, and they had experienced an average of one VOC in the previous year. 19% had experienced chronic pain for more than six months. [2]
About 3% of adult patients always choose gene therapy, and 7% always continued standard care. 2% of parents always chose gene therapy, and about 5% always continued standard care. This suggests that gene therapy was not the automatic choice and that the tradeoff of risks and benefits played a role in their decision-making.
“Both adult patients and parents more often opted for gene therapies offering higher chances of eliminating SCD symptoms and longer life expectancy, and less often opted for gene therapies with higher chances of death, controlling for other factors.”
As you would anticipate, gene therapies that made individuals asymptomatic or improved longevity were always preferred. The tradeoff involved minimizing risk. Of the two risks, death or an increased risk of cancer, cancer was essentially ignored by patients and parents. This suggests that the FDA's concerns about “off-target effects” are not shared by patients or parents. The prospect of dying over the course of a year following treatment was the driver. Whether this was due to its immediacy as compared to the more temporally ambiguous “increase in the lifetime risk of cancer” is a question that remains unanswered.
One of the proposed risks was a 30% chance of dying within the year of initiating gene therapy. This is a massively prohibitive risk. For context, fixing a hip fracture carries an 8.7% risk. But, patients and parents still opted for gene therapy. The researchers conclude that “it is a reflection of the great importance of potentially curative therapies.”
“Disease severity was observed to have a large influence on benefit-risk preferences for gene therapy.”
The tradeoffs were further characterized by whether the adult patient or child currently had mild or moderate symptoms. Adult patients with more symptomatology were more inclined than the less symptomatic to choose gene therapy over standard care. For parents, the mere fact that their child was symptomatic was sufficient to move toward gene therapy. Severity was not a concern. When all participants were told to consider if their symptoms worsened, more moved towards gene therapy, but parents did so to a greater degree than adult patients. As any parent will readily point out, we can tolerate far more “suffering” for ourselves than for our children.
The researchers offered the participants a bargain; they could identify a minimum acceptable benefit for a given risk of death. The results surprised me.
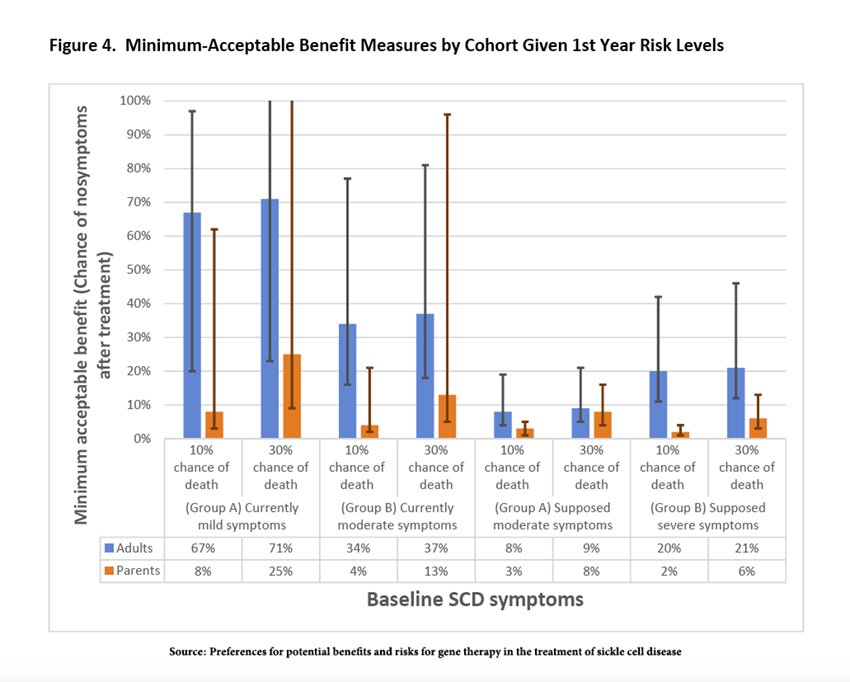
Across the entire spectrum of options, parents exhibited “high levels of risk tolerance” and were willing to accept a much lower minimum acceptable benefit than adult patients. As it turns out, this higher risk tolerance is “consistent with previously documented results.” I am not sure I would make the same choice, but luckily, I am not in a position to make that decision. The less surprising feature of risk tolerance was that those with moderate disease accepted less risk, suggesting, in the words of the researchers, “adaptation as they gain experience with new or more severe symptoms.”
Navigating hope in a hopeless situation involves tradeoffs that we all might make. However, the measure of what is acceptable concerning risks and benefits can vary dramatically from our expectations. We see this in the research presented and the reaction of patient advocacy groups to the glimmer of a successful treatment for sickle cell disease.
[1] A variety of IVF technologies can manage the possibility of infertility.
[2] Parents reported that, on average, their child’s worst year involved 3.8 sickle crises requiring hospitalization.
Source: Preferences for potential benefits and risks for gene therapy in the treatment of sickle cell disease Blood Advances DOI: 10.1182/bloodadvances.2023009680
Tracking the FDA advisory panel on the first CRISPR-based treatment for sickle cell disease STAT

Chuck Dinerstein, MD, MBA
Director of Medicine
Dr. Charles Dinerstein, M.D., MBA, FACS is Director of Medicine at the American Council on Science and Health. He has over 25 years of experience as a vascular surgeon.


