
One of the many horrors that drug discovery chemists constantly wrestle with is called pharmacokinetics (PK) – the science of what the body does to a drug (1). Poor PK properties, for example, rapid metabolism or the lack of absorption from the GI tract into the blood, are one of the common reasons that drugs fail. It can make you crazy.
It turns out that the body can be quite uncooperative, which is rather rude since we're trying to do something to make it feel better. You'd think that the stupid thing might make at least a perfunctory effort to embrace the medicines we're trying to give to it. But no – the ungrateful SOB seems to take great joy in making scientists' lives miserable by finding ways to sabotage the drug that we spent years discovering.
Let's say a group of dorks chemists has spent three years coming up with a potent antiviral drug that kills (2) the virus but not its host cells (3). Better still, when the drug is given to rats and mice intravenously it protects them against infection (prophylaxis) and also treats animals who have already been infected first (therapy). To make its profile even better, the dose of the drug that is needed to prevent/treat the viral infection is far lower than the toxic or lethal doses (a high therapeutic index). Things sure look rosy, right?
Not so fast. When this drug is given orally to the rodents it does nothing. No protection, no treatment, no nothing. Which also means no raise, no bonus, and no promotion. And a manager with breath that could take the heat shields off the space shuttle face berating you in an animated fashion. Life is not good.
But we are not helpless. There are known methods, some of them very clever, that can convert this annoying drug into something that will work when swallowed.
TWO REASONS WHY ORAL DRUGS FAIL
There can a number of explanations for this common, but seriously frustrating screw-up, but usually, it's one of two:
- Metabolism - The drug gets absorbed in the gut (like it should) and travels to the liver via the portal vein. But then the liver decides "Oh boy! This looks yummy" and chews it up into smaller pieces, none of which have any antiviral properties. Or, the liver may decide that it just doesn't like the looks of the drug - the chemical equivalent of racial profiling. So sometimes it attaches a water solubilizing group to it (this process is called conjugation), sending it right to the kidneys which cheerfully expel it in the urine.
- Lack of absorption - In this case, the drug does have the right properties to get absorbed through the intestinal wall (more on that later) and instead, keeps going until it reaches the bottom of the rat cage chemically unchanged, but not in a form that would encourage most of us to take it. Unabsorbed drugs are usually excreted in the feces.
WHY DOES THIS MATTER NOW?
One word - remdesivir. Last week's trial data showed that the drug was moderately effective in helping patients in hospitals recover more quickly and die less frequently. As I wrote (See Remdesivir 1st Controlled Trial Is No Cause For Celebration) remdesivir never got the chance to show what it can do earlier in the infection when it is more likely to be effective (3) because it was given only to patients who had already suffered from a full-fledged viral attack.
This is because remdesivir is administered intravenously, not orally, which limits the public's access to the drug. It is far more time consuming and expensive to give IV drugs at a hospital or infusion center than to swallow a pill.
Most importantly, we are missing the single most important piece of information about the drug– can remdesivir be used early in the infection to make COVID-19 a milder and less dangerous disease? Could it keep people out of hospitals and off ventilators instead of treating them once they're there? Because in the absence of an effective orally administered antiviral drug (or vaccine) there is nothing to stop coronavirus except sitting at home.
WHY CAN'T REMDESIVIR BE MADE INTO A PILL?
Of the two possibilities, I discussed above I'm putting my money on #2 - poor absorption. There are numerous precedents – molecules that are structurally similar to remdesivir – that have had the same issue. But this is a problem that has been solved many times in a variety of different ways. It's a pretty safe bet than remdesivir needs to be chemically modified so that it gets absorbed and then regenerated in the blood. In other words, a pro-drug approach is needed.
WHY WOULD REMDESIVIR NOT BE ABSORBED WHEN GIVEN ORALLY?
Some properties that make drugs more likely to be absorbed include:
- A low molecular weight. Less than 500 is preferable.
- Compactness (a ring instead of a long chain)
- Some solubility in water and some in oil.
Some properties that make a drug less like to be absorbed include:
- A high molecular weight
- Floppiness (a long chain instead of a ring)
- Solubility in water but not in oil
- Solubility oil but not in water
HOW THIS PROBLEM HAS BEEN SOLVED IN OTHER DRUGS
1. N-Hydroxycytidine (NHC) and EIDD-2801
As I wrote in April, one of the many anti-coronavirus candidates being evaluated is called N-hydroxycytidine (NHC). But NHC doesn't work when fed to mice; it is not absorbed in the gut and must be administered by injection. But this property can be altered by a simple chemical transformation - esterification of one of the three hydroxyl groups (Figure 1).
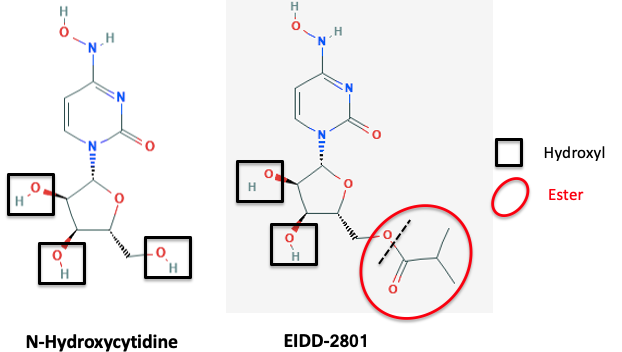
Figure 1. Hydroxyl groups (black squares) are polar, water-soluble, hydrophilic groups. NHC is a ribose-like molecule containing three of them. It is highly water-soluble and poorly soluble in oil. These properties typically result in poor absorption of drugs and also difficulty in penetrating cell membranes, both those in the intestinal wall or the cells which have been infected by the virus. But the conversion of one of these hydroxyl groups to its isobutyrate ester (red oval) results in a more "balanced" (and more drug-like) molecule, which is orally active (a term we use to describe drugs that can be given in pill form).
2. Tenofovir and Tenofovir Disoproxil
Even though tenofovir was approved two decades ago, it remains a critical weapon against HIV/AIDS. But the drug in the pill is not tenofovir itself. Tenofovir contains a phosphonic acid group (4), and there is a better chance of a bassoon making it from your stomach into your bloodstream than a phosphonic acid, which automatically makes a molecule highly water-soluble. A pro-drug (5) approach solved this problem beautifully (Figure 2).
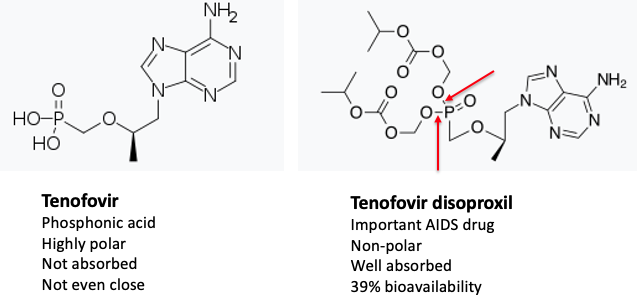
Figure 2. (Left) Molecules like tenofovir, which contain a phosphorous-oxygen double bond, a phosphorous-carbon bond, and two hydroxyl groups belong to a class of molecules called phosphonic acids, which are notoriously awful for penetrating cells. The red arrows indicate the two ester bonds. Ignore the rest of those spaceship-looking things; it's just a dressed-up phosphonate ester. Ref: Science Direct
QUIZ! (for either of you who are still conscious): The red arrows show the phosphonate ester bonds that permit tenofovir to be orally absorbed and also to enter cells. Once inside the cell, the phosphonate ester bonds break, resulting in the release of tenofovir - the active drug (5). What causes those bonds to break? Good luck answering this one. If you get it right I will buy you a Mercedes.
3. Acyclovir and Valtrex
In the first two examples, esterification improved oral absorption of poorly-absorbed drugs by converting polar, hydrophilic drugs into pro-drugs that are less polar and more lipophilic. But pro-drugs can work in a variety of ways, as exemplified by the herpes drug acyclovir and its pro-drug valacyclovir (Valtrex).
Unlike NHC and tenofovir, acyclovir is orally active but not spectacularly so. The drug, which was approved in 1982, is effective; it does pass through the intestinal wall into the blood and then into cells, but it doesn't do so very well Its bioavailibility (6) is 10-20% - not so hot. As shown in the first two examples, oral absorption was improved by a pro-drug approach, in a rather different way. (Figure 3).
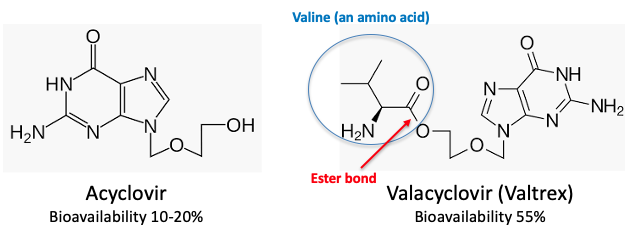
Figure 3. Acyclovir and Valtrex are both used in pill form to control herpes, but Valtrex is a superior drug because of the valine ester (blue circle). The valine ester-molecule "looks" enough like the amino acid valine to be taken up by amino acid transporters and "carried" into cells.
A few things worth noting. Unlike NHC and tenofovir, acyclovir has only one hydroxyl group, so it has "looks" like it will have better drug-like properties than those other two monstrosities. It does. As a result, acyclovir is orally active – enough so to be sold in pill form – but it needs to be taken 4-5 times per day and a lot of it (80-90%) is wasted – it doesn't get into the bloodstream. The solution here is also the esterification of the one hydroxyl group, but it works very differently than in the first two examples. Valtrex is more polar (and water-soluble than acyclovir, so what the hell is going on?
It's the valine ester. Valine is one of the 20 amino acids, the building blocks of protein, that is produced by DNA by humans. (Proteins are large chains (>50 amino acids hooked together. Peptides are smaller (2-50 amino acids). When proteins are degraded in the stomach they form amino acids and also small peptides. Neither the peptides or amino acids are particularly well-suited to be absorbed through in the gut, but we'd be rather dead without them. So the body came up with a near trick - amino acid transporters. These transporters grab onto amino acids and force them through the stomach and intestine.
The transporters are quite efficient, but they're not terribly bright. Valacyclovir is neither an amino acid nor a peptide; it's just a drug that contains a valine piece. But this is sufficient to trick the stupid transporters into "thinking" that the drug is an amino acid, so it actively transports the drug into the blood. Then the valine ester then is chopped off by enzymes in the blood called esterases, and now there's a whole bunch of acyclovir in the blood. The significant increase in bioavailability is reflected by the improved dosing schedule (1-2 times/day vs. 4-5 for acyclovir). Hooking amino esters to poorly absorbed drugs gives us another pro-drug approach to make them orally active.
4. Can these techniques be applied to remdesivir?
Gilead knows a bazillion times more about these things than I do, so if they are not doing this (7) there must be a good reason, either something in the works or the realization that remdesivir isn't a good enough antiviral drug or already have a solution in the works. A pro-drug of remdesivir should be able to make it orally active; the molecule has already been partially "decorated" in this way (Figure 4).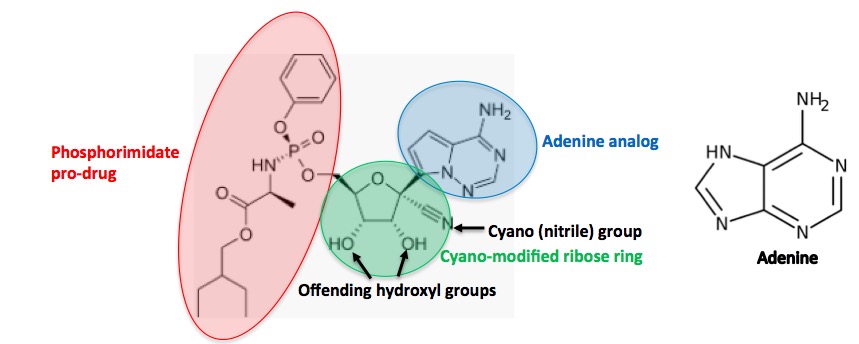
Figure 4. Remdesivir can be divided into three fragments. The phosphorimidate (red, good luck using this in Scrabble) is a well-known pro-drug of phosphonic acids called Pro-tide. It enables drugs to be absorbed into the blood and then permeate cell membranes, where it is broken down to give the active drug (5). The ribose fragment (green) contains a (rare) cyano group. The blue circle contains a "mixed up" adenine molecule, which is critical for inhibiting the RNA polymerase enzyme that the coronavirus requires for replication.
So, why is remdesivir not orally active? It (almost) has to be the two hydroxyl groups, which, as in the case of NHC (has 3 of them) became orally active by masking one of them as an ester. Would this approach work with remdesiviir? Ask Gilead. They surely know.
5. Now what?
I cannot possibly overemphasize the importance of an effective and widely-available coronavirus pill. This would be a game-changer, For antiviral drugs to work best, it is critically important that they are taken early in the infection - before the virus has explosively replicated and done its damage.
I'm not sure what Gilead's plans are but Vivtex, a Cambridge-based biotech, which was founded by MIT's Robert Langer, is investigating new formulations that may help make poorly-absorbed drugs (and biologics) orally active. The company is applying its technology to a number of drugs, including remdesivir. We should all wish them well.
A more straightforward (and obvious) approach would be to chemically modify one or both of the hydroxyl groups using methods such as I described earlier. If this is being done no one is talking yet. I like this approach because it has a track record. It should work.
Then again, we shouldn't have to be running around wearing masks. These are strange times. Rules no longer apply. And there is still much to be learned.
NOTES:
(1) Pharmacodynamics is the "opposite" of pharmacokinetics. It is the study of what the drug does to the body.
(2) Technically, you cannot "kill" a virus because it is not alive. Inactivate would be more accurate.
(3) Viruses (some of them) can be grown in a monolayer of cells in a small well. It is easy to determine if a given chemical compound inhibits the growth of the virus. This process is done by robotics. Millions of unique compounds can be tested in a short time. The catch - viruses only replicate in health host cells. If the compound inhibits viral growth but is also toxic to the host cells it is not an antiviral drug, just a toxin.
(4) The similarity of carboxylic acids and phosphonic acids and their esters.
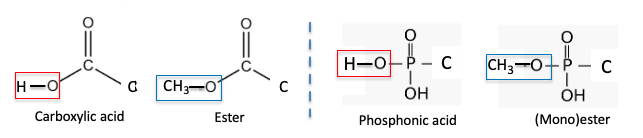
(5) It's actually more complicated than this. Cells add two phosphate groups to the phosphonic acid. This is why it is sufficient to get the phosphonic acid into the cell. Don't ask.
(6) Bioavailablity is the measure of how much of a drug gets into the blood when taken orally vs. given IV. The higher the number the better.
(7) Gilead has stated that they have no plans to develop a pill form of remdesivir.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


