
Hiroshima and Nagasaki
There was clearly an increase in cancer incidence among survivors of the initial blast, prompting a registry of those 44,000. Initially, after roughly two years, there was a wave of leukemias, predominantly among children, reflecting the faster cell growth of blood elements and children in general. Later, at about ten years, a wave of solid tumors, e.g., colon, breast, thyroid, now mostly among adults, reflecting both the slow turnover of cells within our solid organs and the effects of age on our defenses against cancer. The elevated relative risk of leukemia among survivors was 16%; for solid tumors, 10%. Why not more?
As you might expect, the incidence of cancer increased with exposure to radiation as measured in Gy [1]. Here are the plots for elevated risk and dosage.
 Look carefully at the “dose-response” at the lower exposures, close to zero. The dose and response are linear for the solid tumors (on the left). But for those leukemias (on the right), while the incidence remains elevated, at about a 6% increase,
Look carefully at the “dose-response” at the lower exposures, close to zero. The dose and response are linear for the solid tumors (on the left). But for those leukemias (on the right), while the incidence remains elevated, at about a 6% increase,
“In contrast to dose-response patterns for other cancers, that for leukemia appears to be nonlinear; low doses may be less effective than would be predicted by a simple linear dose-response.”
There appears to be a threshold below which exposure to radiation does not increase your risk of developing cancer. Why might that be the case?
The EPA has based its regulations of chemical carcinogens on the solid tumor model of mutation to cancer from radiation, termed the linear non-threshold model. We have learned a great deal about how cancers developed in the last 70 years, much of which would suggest that the leukemia model, where a threshold exists, more accurately describes the effect of low-dose carcinogens. Let’s take a deeper contextual dive.
Carcinogenesis is a multi-stage process.
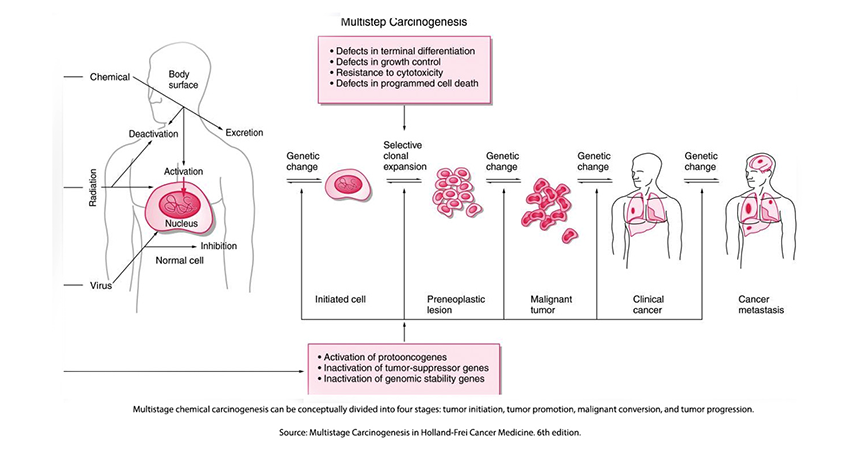 The effects of radiation on the citizens of Hiroshima and Nagasaki spurred scientific inquiry into the underlying mechanisms of human carcinogenesis – the development of cancer. The radioactive model suggested that a single radioactively induced alteration in DNA, a mutation, was irreversible, ultimately resulting in cells that multiplied without limit, cancers. This early model or hypothesis has proven too simplistic; carcinogenesis in biological systems proceeds through several phases.
The effects of radiation on the citizens of Hiroshima and Nagasaki spurred scientific inquiry into the underlying mechanisms of human carcinogenesis – the development of cancer. The radioactive model suggested that a single radioactively induced alteration in DNA, a mutation, was irreversible, ultimately resulting in cells that multiplied without limit, cancers. This early model or hypothesis has proven too simplistic; carcinogenesis in biological systems proceeds through several phases.
The initiation of cancer begins with a mutation in DNA. For radiation, it may be a one-and-done process, altering a DNA base and changing the code. For chemical carcinogens, the carcinogen binds to the DNA forming a “DNA-adduct,” which, like radiation, alters the DNA code. There is little evidence that chemical carcinogens initiate cancers without the presence of DNA adducts; in fact, these adducts are a biomarker of chemically induced cancers. But while necessary, alterations in our DNA code are not sufficient to result in cancers.
The promotion of tumors requires cells to replicate; the greater the replication, the more common a “cancerous” mutation. The promoters of replication increase the number of cells “at risk for malignant conversion. Promoters are not necessarily abnormal; they may simply be how quickly these cells typically replicate. For example, the lifespan of a cell lining our intestine is about ten days; for those fat cells, years. Promotion reduces the time from initiation to the formation of a cancer – the latency or lag period. Inflammation is a promoter, reducing latency; often, continued exposure to the initiating chemical acts as a promoter.
The presence of promoters increases the relatively low probability of malignant conversion or progression. This conversion to a cell without limits may be impacted by mutations that affect protooncogenes and tumor suppressor genes. Mutations in these normally present genes “confer to the cells a growth advantage as well as the capacity for regional invasion, and ultimately, distant metastatic spread.” The accumulation of these mutations seems to be the biomarker of progression.
There are biological reparative defenses, including our ability to repair DNA mutations. Repair was never a consideration in the irreversible changes of radiation. Coupled with our greater understanding of the biological underpinnings of carcinogenesis rather than simply a physics-derived model, it is clear that cancer development in the presence of a chemical cause has a wide variability – it is not one and done. The variability is so great that there is a school of thought that cancers are more “Bad luck” from random and then aligning events than from specific exposures.
Once again, when we consider our biological foundations, they are complex, messy interactions, even though they are rooted in physics “laws,” which tend to be more precise or clear. (At least until you consider quantum physics). Biological carcinogenesis is the interaction of not just the dose but the properties of the carcinogen and “the various endogenous and exogenous factors affecting the organism.”
“…given any carcinogen and any organism under any set of conditions, the general tenet is that high doses shorten latency and appear more linear-like while low doses lengthen latency and appear more threshold-like.”
The Big Problem with Small Numbers
To detect differences between two groups, say those exposed to a carcinogen and those not, one must ensure a sample size that has sufficient “statistical power” to “correctly reject a false null hypothesis (e.g., the means of two distributions are equal) – that in this case, the carcinogen results in more cases of cancer in those exposed.
Without belaboring the mathematics, when a carcinogen is present at a very low concentration causing very few but slightly more cancers than may happen by chance, the sample size must be very large – a slight difference requires a large sample. The sample size must also be increased when you want to be more “statistically certain” about your finding of an effect, and the sample size needs to be further increased when there is some variability built into the outcome. From a biological view, carcinogenesis has great variability, requiring initiation, a promoter, and progression.
If a large sample size was not enough, carcinogenic effects in humans frequently take years to develop. Radiation exposure does not reveal its harmful consequences for 5 to 10 years; for chemicals, we may be considering decades. As a result of the logistical problems of thousands of patients followed for years, if not decades, we have no studies looking at the effects of low doses of chemicals on the development of cancers. We do have data on the impact of large chemical exposures in animals and observational data in humans. The observational data on smoking was the deciding factor in the 1964 Surgeon General report on the harms of cigarettes.
In the absence of randomized controlled studies, we have taken the data on high exposure to radiation and projected, extrapolated, what the impact of lower exposures will be.
“What dose-response model can describe and predict responses of ionizing radiation and toxic agents in the low dose zone?”
Here are the three models or extrapolations of high-dose exposure to radiation as a carcinogen. Each seeks to describe how the cancer risk changes with lower doses where we have no information. For the sake of this discussion, we are just looking at the bottom left of a larger graph – just the unknowns of low exposure.
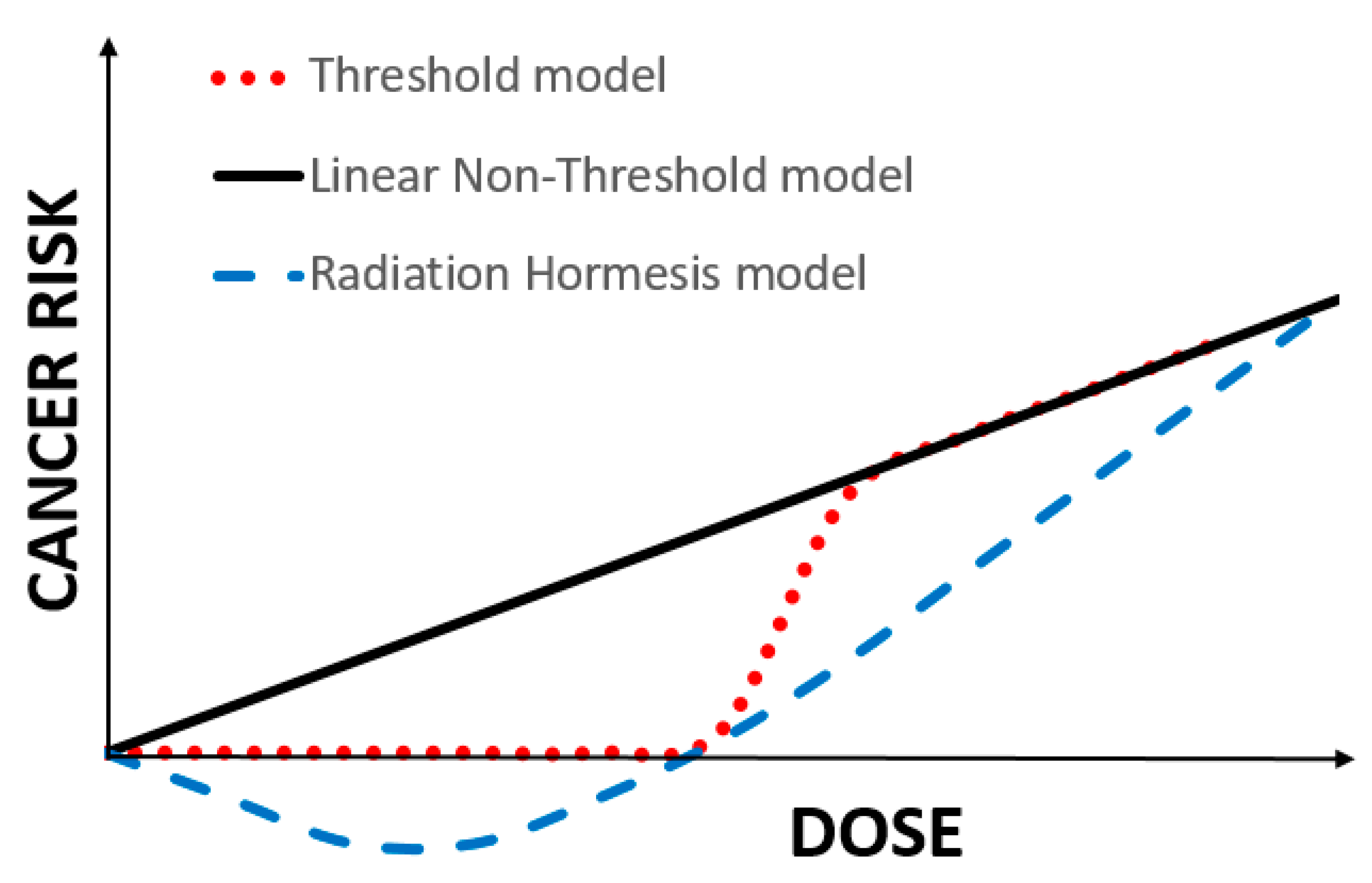 The linear non-threshold model (LNT) has been the regulatory standard for over half a century based on the effects of ionizing radiation and assuming that the mutations from radiation “were cumulative, non-repairable and irreversible.” In time, this extrapolation was extended to chemical carcinogens
The linear non-threshold model (LNT) has been the regulatory standard for over half a century based on the effects of ionizing radiation and assuming that the mutations from radiation “were cumulative, non-repairable and irreversible.” In time, this extrapolation was extended to chemical carcinogens
“It is worth emphasizing that one molecule of a mutagen is enough to cause a mutation and that if a large population is exposed to a ‘weak’ mutagen, it may still be a hazard to the human germ line, since no repair system is completely effective, there may be no such thing as a completely safe dose of a mutagen.”
– Bruce Ames [2]
A threshold or dose-response model had existed in the biological sciences for many years, annunciated by Paracelsus, "All things are poison, and nothing is without poison; the dosage alone makes it so a thing is not a poison." Others suggested that very small doses of chemicals might have a salutary effect giving way to more adverse and lethal consequences as the dosage increased – the hermetic model.
Our understanding of biological knowledge, especially concerning carcinogenesis, has dramatically expanded from those days at the end of World War II when a radiation model of cancer was apparent. In addition to the alignments necessary for the initiation, promotion, and progression of a tumor, there are a host of facts influencing whether oral or respiratory exposure to a carcinogen results in the opportunity for initiation. The internal exposure to a carcinogen from its external sources is mediated by our physiological response to
- Absorption
- Distribution
- Metabolism
- Storage
- Excretion
All of these bear on the number of carcinogenic molecules reaching a DNA target, their capacity in any one instance to result in mutation, and our momentary ability to repair the injured cell. Taken together, it is more biologically plausible that carcinogenesis is not one and done as in radiation-induced mutation. It may fail more often than it succeeds, creating a dynamic biological threshold for which dose or exposure is only one of many factors.
“Thus, the capacity of a cell to repair DNA damage before the damage can affect permanent carcinogenic alterations produces a “threshold” response that depends on both the cellular capacity to repair DNA and the rate at which the repair occurs.”
There are multiple objections to why a threshold model is less accurate than the LNT model. Some may be easily dismissed with the additional knowledge of the last 75 years. While a single or a few molecules can indeed result in mutation, that transformation is not irreversible or irreversibly propagated. It takes more than a mutation in a few cells to transform into cancer. Radiation and chemical-induced mutations share similar but not identical pathways.
The strongest objection lies in the heterogeneity of our individual biological responses. While we each may well have a threshold for a specific substance initiating carcinogenesis, when we aggregate as a population, that threshold may no longer be apparent. A host of social and cultural factors, like stress, diet, access to medical care, and cumulative vs. acute exposure through work or home, may confound the ability of epidemiology to identify a population's “threshold.”
The hormetic model where small doses of a substance, radiation, or chemical may have a salutary effect is a variation of the threshold model. As with the other two models, there is no randomized controlled study that proves its value over the others. There are other biological reasons to consider its underlying truth. Most of the primary measures of our physiology, sodium, potassium, glucose, and lipids, all have “Goldilocks” values; neither too high nor too low. When concentrations are plotted against our health, all form that hermetic “Nike swish.” It even holds for UV radiation, with small amounts necessary to promote the physiologic requirements to produce Vitamin D, and larger and larger amounts resulting in sunburn or even skin cancers.
All of these models, LNT, threshold, and hermetic give us a very accurate picture of carcinogenesis at high levels – they are essentially identical. But it is at the very low doses, at the regulatory doses, that the models diverge. With no scientific certainty, what is the best model to protect the public good?
Enter the EPA
The initial focus of the Environmental Protection Agency was environmental pollutants and their adverse health effects, primarily environmentally induced cancers. The radiation-induced model of carcinogenesis captured and studied since the 1950s in the LNT model was a good fit with the leadership beliefs of a mutagen mediating a carcinogenic response in proportion to its dosage. While the LNT’s, one-and-done model, might be theoretically a good approach to protecting the population, it quickly became apparent that it was not an economically viable option. [4]
As to be expected, regulated industry, both chemical and radiologic, favored an equally and more plausible threshold model for carcinogenesis. This would reduce their costs. The inability of epidemiologic studies to indicate which of the two models was more accurate at the low dosage levels left regulatory “wiggle-room” in establishing the scientific basis for regulation. In the face of uncertainty, the precautionary principle applies.
“..if a product, an action, or a policy has a suspected risk of causing harm to the public or to the environment, protective action should be supported before there is complete scientific proof of a risk.”
The preeminence given to the LNT hypothesis resulted in analytical testing that would suit the requirements of LNT’s extrapolations. This includes the choice of experimental animals and the number, spacing, and dosages. There was no need for long-term studies of multiple low doses. A few data points below the minimum lethal dose [3] would be “sufficient” to extrapolate into the regulated low-dose range. In this range, the LNT model diverges from the threshold model.
From a practical point of view, strict regulation using the LNT model would shut down significant portions of our energy and food economies. The EPA adopted a de minimus standard, an arbitrary point on that LNT extrapolation, a risk of 1 to 1.5 cancers per 1,000,000 individuals over a 70 to 80 year lifetime. Here is the EPA’s disclaimer of their de minimus standard
“The URE [unit risk estimate] and CPS [carcinogenic potency slope – the LNT] are plausible upper-bound estimates of the risk (i.e., the actual risk is likely to be lower but may be greater). However, because the URE and CPS reflect unquantifiable assumptions about effects at low doses, their upper bounds are not true statistical confidence limits.” [emphasis added]
[1] 1 Gy (gray) is equivalent to 1000 millisieverts or 125 abdominal CT scans, 10,000 chest x-rays, or the background radiation from living on earth for about 273 years.
[2] Dr. Bruce Ames is an icon in environmental and mutagenic research. He developed a rapid test to ascertain the mutagenicity of chemical agents; it remains a gold standard. As his understanding of carcinogenesis has evolved, and has “argued that environmental exposure to manufactured chemicals may be of limited relevance to human cancer, even when such chemicals are mutagenic in an Ames test and carcinogenic in rodent assays.”
[3] This might be a foretaste of the argument over masks, social distancing, lockdowns, and shutting down the economy during the time of COVID.
[4] The lethal minimum dose (LMD) is the smallest amount of chemical or radiation to result in the death of test animals. At a higher level, the LD50 reflects the dosage necessary to kill 50% of the experimental animals.
Sources: Multistage Carcinogenesis Holland-Frei Cancer Medicine. 6th edition.
Linear non-threshold (LNT) fails numerous toxicological stress tests: Implications for continued policy use Chemico-Biological Interactions DOI: 10.1016/j.cbi.2022.110064
Focus on Data: Statistical Design of Experiments and Sample Size Selection Using Power Analysis Investigative Ophthalmologic and Visual Sciences DOI: 10.1167/iovs.61.8.11
Risk of Leukemia among Atomic Bomb Survivors and Risk of Solid Tumors among Atomic Bomb Survivors Radiation Effects Research Foundation

Chuck Dinerstein, MD, MBA
Director of Medicine
Dr. Charles Dinerstein, M.D., MBA, FACS is Director of Medicine at the American Council on Science and Health. He has over 25 years of experience as a vascular surgeon.


