
There's a recent, wildly enthusiastic YouTube video that touts the discovery of a new antibiotic that treats various resistant organisms. It's called manikomycin, and it can potentially treat infections from a variety of serious bugs that no longer respond to conventional antibiotics. The need for drugs that treat serious infections like MRSA is acute, so this is clearly a topic of interest.
But, for those of you who were never involved in drug discovery, especially in antibiotic discovery, the video might make you think the new drug—a potentially major advance in the treatment of antibiotic-resistant infectious diseases—is a slam dunk.
Both of us have been involved in exactly this type of research and have learned (the hard way) that slam dunks are few and far between. Let's go back in time for some historical context.
In the 1990s, MRSA (methicillin-resistant Staphylococcus aureus) was becoming an increasingly serious problem. What had once been primarily a hospital-acquired pathogen was beginning to spread into the broader community. These strains were not only resistant to multiple antibiotics but were often highly virulent as well. Serious skin and soft tissue infections became more common, and in some cases, healthy young people—including athletes—developed life-threatening disease. Some died.
At the time, treatment options were severely limited. Vancomycin was essentially the only approved drug that could be relied upon to treat serious MRSA infections. But vancomycin was hardly an ideal solution. It had to be administered intravenously, making outpatient treatment difficult or impossible in many cases. Its efficacy was often less than optimal, particularly in deep-seated infections, and clinicians had to monitor patients carefully because of the risk of kidney toxicity and other adverse effects.
As MRSA continued to spread, the need for new antibiotics became increasingly urgent. Pharmaceutical companies, including Wyeth, launched major discovery programs in search of compounds that could safely and effectively treat resistant Gram-positive pathogens. The scientific challenge was formidable, but so was the medical need.
In search of better drugs
At Wyeth, we initiated what we called a “lookback” program. The company had a vast collection of compounds isolated from living organisms—natural products—many of which had been discovered decades earlier and then set aside for one reason or another. Some dated back to the 1940s. We went back through this collection, asking a simple question: had something useful been overlooked?
To our surprise, the answer was yes.
We found one: AC-98 (Figure 1).
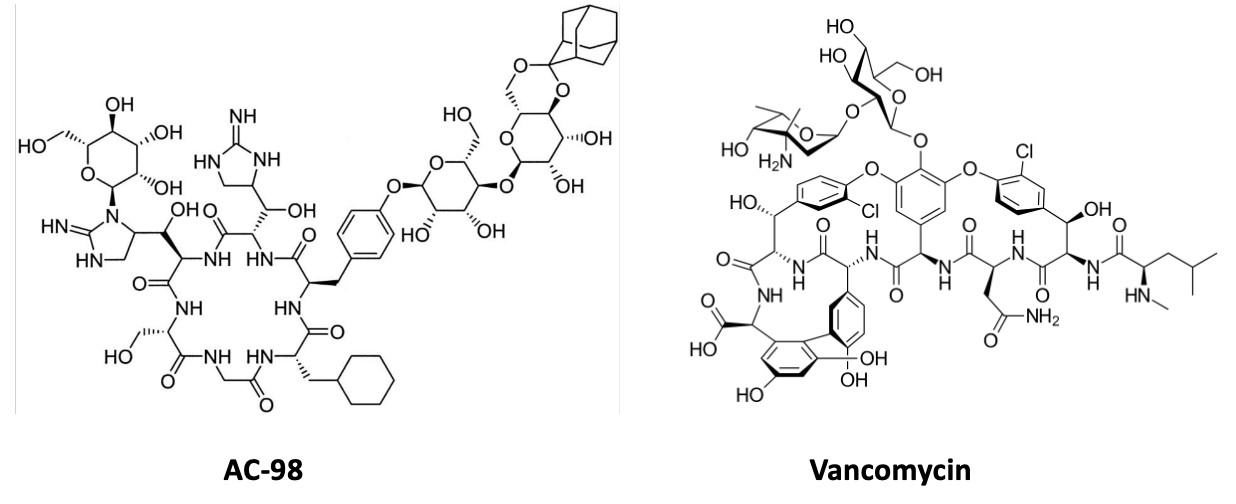
Figure 1. Two chemistry nightmares, AC-98 and vancomycin. Pick your poison.
Reality intervenes
AC-98 looked extremely promising. It was active against a wide range of resistant staphylococci, including strains resistant to both penicillins and vancomycin, because it targeted bacterial cell-wall synthesis in a novel way. But it also shared an important characteristic with vancomycin: it was a very large molecule. Large molecules often make poor drugs because they are poorly absorbed, distribute imperfectly into tissues, and almost always require intravenous administration. Those limitations do not make success impossible—vancomycin itself is proof of that—but they do create additional hurdles. We ran into plenty of them.
As the program progressed, reality set in. We had to develop methods to produce enough material for testing, improve potency through medicinal chemistry, and evaluate safety. Each challenge was met, only to reveal another. Compounds with improved antibacterial activity caused kidney toxicity in animals. New analogs eliminated the kidney toxicity but produced serious blood-clotting abnormalities. After years of effort and substantial investment, AC-98 and its descendants joined the graveyard of antibiotic candidates that seemed destined for success but failed before reaching patients.
Does manikomycin face the same hurdles?
None of this means that manikomycin is destined for the same fate as AC-98. In fact, the new antibiotic brings something to the table that AC-98 never did: a completely new target.
The researchers found that manikomycin binds to a previously unexplored site on the bacterial ribosome, the molecular machine that makes proteins. This so-called E-site has never before been targeted by an antibacterial drug. That alone makes the discovery noteworthy. Drug hunters are often willing to tolerate an ugly molecule (and this one is plenty ugly) if it reveals a beautiful target.
Manikomycin raises some familiar concerns regarding drug development, particularly antibiotic development.
The compound clearly kills bacteria, including some problematic Gram-negative pathogens, but the concentrations required are not breathtaking by modern antibiotic standards. For example, the minimum inhibitory concentrations (MICs), the lowest concentrations needed to stop bacterial growth in the laboratory, were roughly 32 and 16 μg/mL against E. coli and Klebsiella pneumoniae, respectively.
These numbers aren't great because, in general, blood and tissue drug concentrations must exceed the MIC for the drug to be effective. The higher the MIC, the harder it can be to achieve effective drug levels in patients without running into pharmacokinetic or toxicity problems. MICs in the single digits would be more encouraging.
The second concern becomes obvious the moment one looks at the structure. Manikomycin is a giant molecule, weighing in at more than 1,100 daltons and carrying multiple positively charged groups. Medicinal chemists tend to start sweating when they see structures like this. Large, highly charged molecules often make for difficult drugs. They can be challenging to manufacture, difficult to formulate, and prone to pharmacokinetic problems (see below). Getting enough of the compound to the site of infection can be easier said than done.
Which brings us to the third concern: pharmacokinetics – the action of the body on the drug.
The authors report that manikomycin is stable in plasma, but also found that it was cleared rapidly from mice, with a terminal half-life of only about 36 minutes (this is poor). Even more concerning, peak blood concentrations after dosing were relatively modest. In fact, initial mouse infection studies failed to show efficacy, which the authors attribute to inadequate drug exposure, probably arising from poor PK, rather than a lack of antibacterial activity.
Anyone who has spent time in antibiotic discovery has learned the same lesson repeatedly: killing bacteria in a petri dish is the easy part. The real challenge begins when the compound encounters a mammal.
None of this diminishes the importance of the discovery. The identification of a new ribosomal target is a genuine scientific advance. Even if manikomycin itself never becomes a medicine, the work demonstrates that the bacterial E-site can be drugged. Medicinal chemists may eventually be able to exploit that vulnerability using compounds that are smaller, more potent, and more pharmacologically "friendly".
In other words, the most important product of this research may not be manikomycin itself. It may be the door that manikomycin has opened.
Pretty much like the rest of drug discovery research.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


