Sometimes it feels as if it’s March 2020 again. Back then I complained that bureaucratic red tape from the Food and Drug Administration (FDA) and Centers for Disease Control and Prevention (CDC) made Americans wait too long to access COVID-19 tests when they were already widely available in much of the developed world. That made it more difficult to slow the spread of the virus at a time when vaccines and therapeutics were not yet available. Now it appears the FDA and CDC are about to repeat the same mistakes with monkeypox testing and treatment.
In the past, monkeypox outbreaks were rather uncommon, small, and short‐lived outside of the African continent where the virus originated. Because of this, commercial tests have not yet been developed in the U.S. or elsewhere. Increased global travel and trade means that monkeypox will likely become endemic worldwide, increasing the demand for rapid and accurate tests.
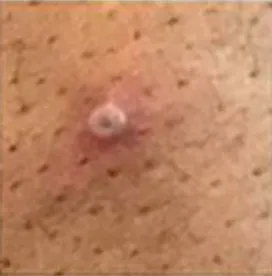
Testing for monkeypox requires sampling secretions from the monkeypox lesions. As in the early days of the COVID pandemic, tests were initially only done at the CDC and some of its 67 designated federal laboratories in 48 states. In late June the CDC authorized Labcorp, Quest, Mayo Clinic, and Aegis Science labs to perform the tests as well. Unfortunately, those labs can detect the presence of an orthopox virus (monkeypox, smallpox, and cowpox are members of that virus family), but positive samples must then be sent to the CDC for typing to confirm they are monkeypox. Several days can go by before confirming a patient has monkeypox. Last month Stanford University School of Medicine announced its Clinical Virology Laboratory developed an in‐house PCR test that bypasses the cumbersome CDC confirmation system and yields a specific result in 24–48 hours. Hopefully, the FDA will not repeat the mistakes of March 2020 by throwing regulatory sand in the gears of academic and commercial laboratories that are responding to the demand for rapid monkeypox tests.
Most monkeypox patients get better on their own without treatment in 2 to 4 weeks. This clade of monkeypox appears non‐lethal. No deaths have occurred anywhere in the world since the outbreak began. But sometimes lesions can become secondarily infected by bacteria and lead to pneumonia, eye infections, and encephalitis. Therefore, it would be good to have a drug that can make monkeypox infections resolve more quickly, before complications develop. Such a drug exists. The antiviral drug tecovirimat or TPOXX, used to treat smallpox and cowpox, works on monkeypox as well. The European Medicines Agency authorized the drug to treat monkeypox in the spring.
Unsurprisingly, regulators in the U.S. are behind the curve. While approved to treat smallpox, the drug is not yet approved to treat monkeypox. But in mid‐July, the CDC began an Expanded Access‐Investigational New Drug (EA-IND) program, through which clinicians can use the drug to treat their monkeypox patients. Under its EA-IND umbrella, the CDC provides TPOXX from the national stockpile through state and local health departments. Initially clinicians had to submit cumbersome paperwork to prescribe the drug. Hoping to ease that burden, the CDC later began allowing clinicians to submit the paperwork after beginning treatment. However, the inconvenience—and disincentive to prescribing—remains. Furthermore, the CDC recommends against prescribing TPOXX until testing confirms a monkeypox infection. Because waiting for the CDC to confirm that a positive orthopox test from one of the commercial labs is indeed monkeypox delays treatment even longer, the CDC lets clinicians treat their patients as soon as they receive a positive test for orthopox—since it is highly likely viral typing will confirm it is monkeypox.
Like the antiviral Paxlovid used to treat COVID, TPOXX must be started soon after symptoms appear for it to work to resolve the infection and heal the lesions quickly. When the FDA issued Emergency Use Authorization (EUA) of Paxlovid on December 22, 2021, the agency included unworkable prescribing guidelines and required a positive test before Paxlovid could be prescribed to patients. At the time, rapid at‐home self‐administered test were not widely available, and in most cases, patients had to wait 2 to 3 days for a test result and then get in to a doctor’s office. By then the window for the drug to work might have already closed. Josh Bloom and I argued in the New York Daily News back then that these regulations were counterproductive and harmful to COVID patients. We concluded:
A better approach would be for the FDA to remove the testing requirement, stop micromanaging nuanced clinical decision‐making, and let doctors use their knowledge and judgment in deciding to whom and when to recommend the drugs. Physicians, not government agencies, are best equipped to be making critical decisions concerning the health of their patients.
Eventually, as rapid COVID tests became widely available, the Biden Administration implemented the “Test to Treat Program,” a much more efficient strategy, which gives patients ready access to rapid tests in retail pharmacies and wisely permits pharmacists to prescribe Paxlovid to patients who test positive.
Alas the FDA and CDC seem poised to repeat with monkeypox many of the mistakes in testing and treating that they made with COVID. In contrast, the New York City Department of Health states in its Interim Guidance for Treatment of Monkeypox:
Tecovirimat should be considered broadly for treatment of monkeypox, and patient selection is at the discretion of the treating clinician under the EA-IND. Both oral and intravenous formulations are available. Empiric treatment can be considered if there is appropriate clinical indication prior to laboratory confirmation, especially in the context of limited or delayed testing. (emphasis added)
Unfortunately, New York’s Department of Health can’t unburden prescribers of the CDC’s required paperwork. But at least one public health agency is showing signs it is learning from the mistakes of the recent past.
#Reprinted with permission from the Cato Institute, Dr. Singer's original post can be found here.




{kind=link}